Jekyll2024-07-23T15:44:21+00:00https://ucsdml.github.io//feed.xmlUCSD Machine Learning GroupResearch updates from the UCSD community, with a focus on machine learning, data science, and applied algorithms.FairProof : Confidential and Certifiable Fairness for Neural Networks2024-07-23T00:00:00+00:002024-07-23T00:00:00+00:00https://ucsdml.github.io//2024/07/23/fairproof
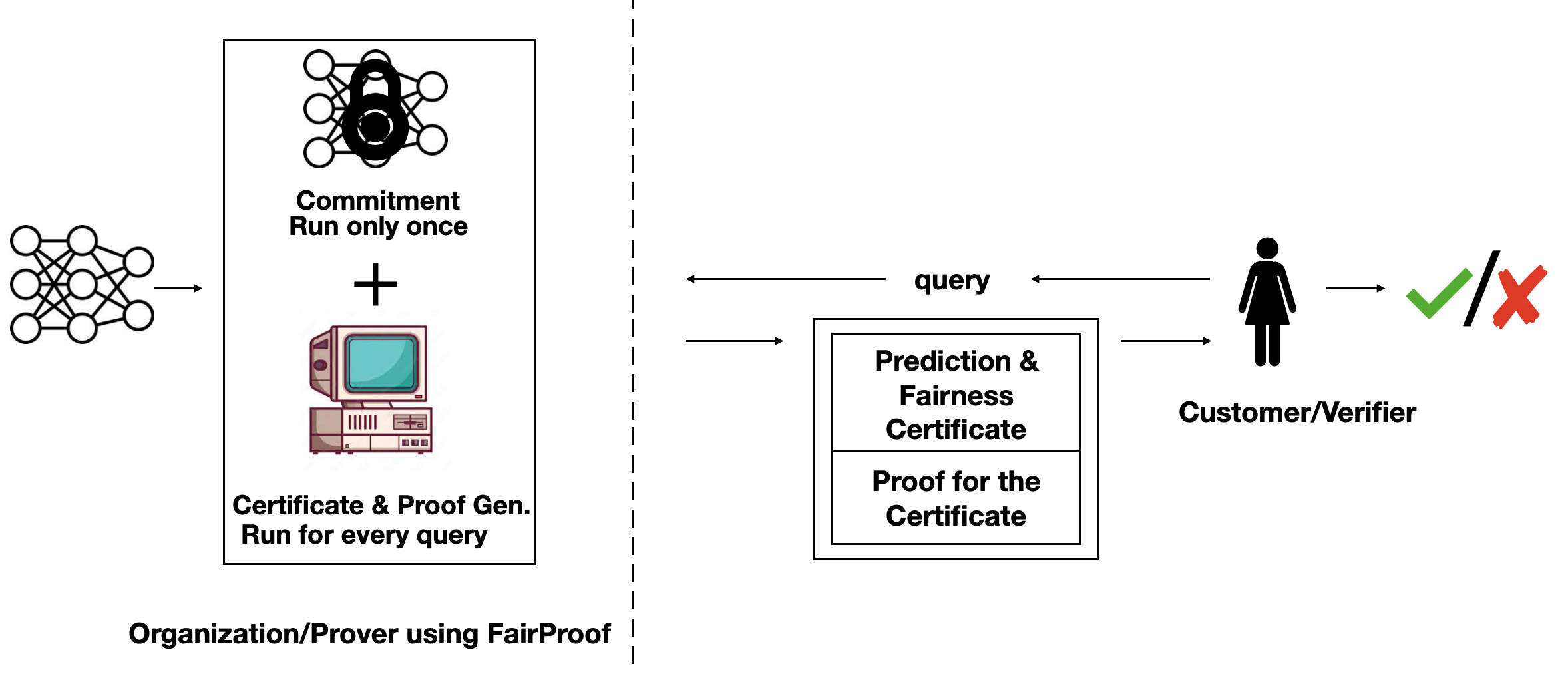
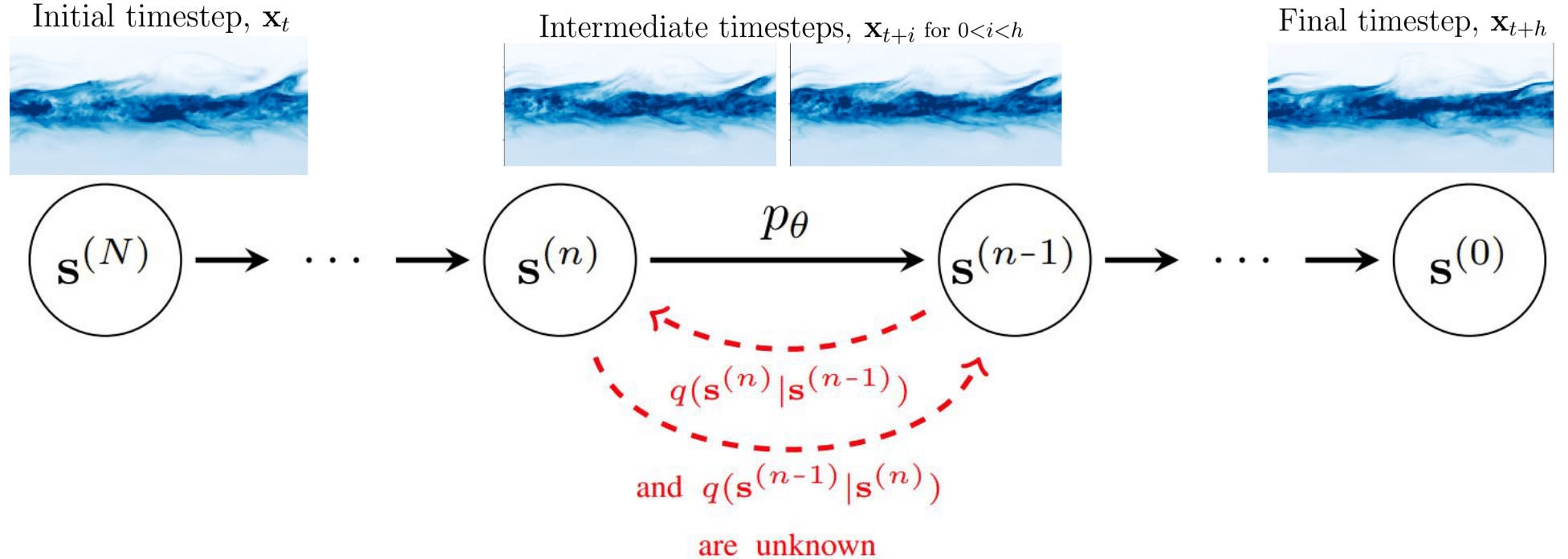
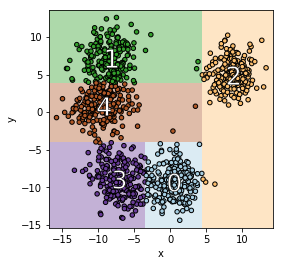
Setting of FairProof. Customer (which is also the verifier) submits a loan application to a bank (which is the prover). The bank uses a machine learning model to make decisions and responds with a decision, fairness certificate and a proof of the correct computation of this certificate. This proof is verified by customer.
Introduction
As machine learning models proliferate in societal applications , it is important to verify that they possess desirable properties such as accuracy, fairness and privacy. Additionally due to legal and IP reasons, models are kept confidential and therefore verification techniques should respect this confidentiality. This brings us to the question of how do we verify properties of a model while maintaining confidentiality.
The canonical approach to verification is ‘third-party auditing’ , wherein an external auditor uses API queries to estimate the value of the property . However, this approach has certain problems including 1) use of auditing datasets which can be manipulated 2) model swapping wherein the model is changed post-audit 3) leaking the model in the process, which loses confidentiality .
Our Key Idea
Motivated by these issues, we propose an alternate framework for verification which is based on Zero-Knowledge Proofs (ZKPs) , a cryptographic primitive. To put it simply, a ZKP system involves two parties, a Prover and a Verifier. Prover proves the correct computation of an algorithm which uses model weights while Verifier verifies the proof given by the prover without looking at the model weights.
Guarantees of FairProof
In this work, we propose a ZKP system called FairProof to verify the fairness of a model. We want FairProof to give the following three guarantees: 1) ensure that the same model is used for all customers, 2) maintain the confidentiality of the model and 3) the fairness score is correctly computed without manipulation using the fixed model weights only. The first requirement is guaranteed through model commitments – a cryptographic commitment to the model weights binds the organization to those weights publicly while maintaining confidentiality of the weights and has been widely studied in the ML security literature . The other two requirements are guaranteed with ZKPs.
Problem Setting
Consider the following problem setting: on one hand, we have a customer which applies for a loan (i.e. query) and corresponds to the verifier while on the other, we have a bank which uses a machine learning model to make loan decisions and corresponds to the prover. Along with the loan decision, the bank also gives a fairness certificate and a cryptographic proof proving the correct computation of this certificate. The verifier verifies this proof without looking at the model weights.
Two parts of FairProof
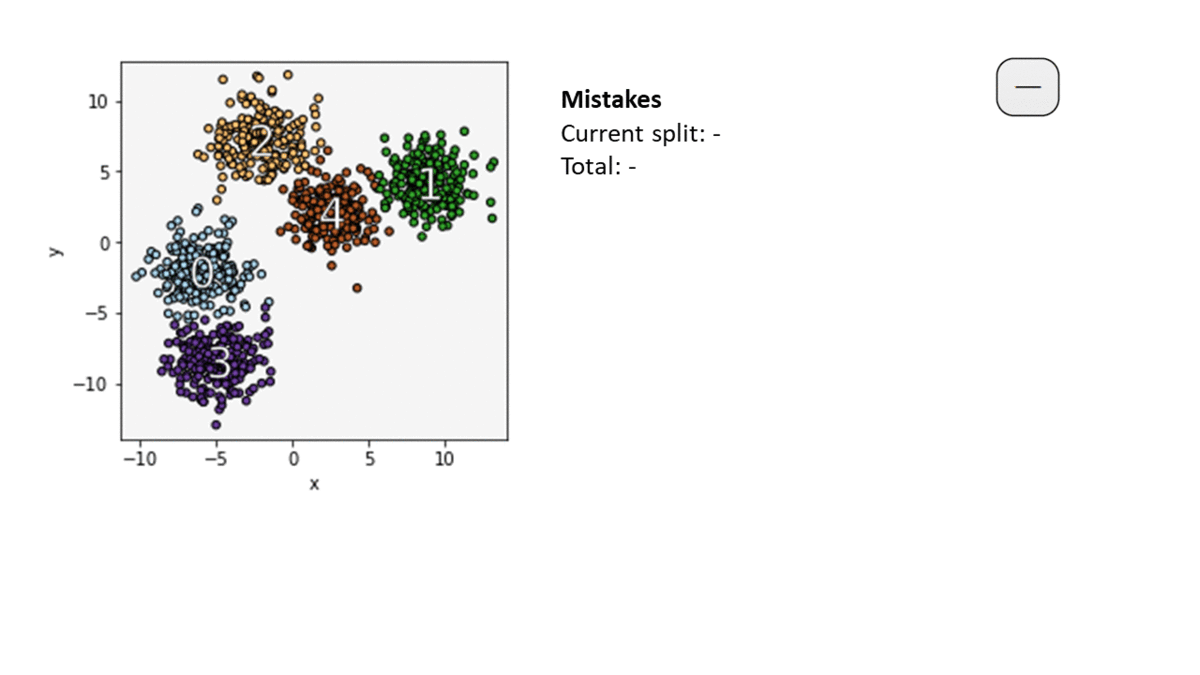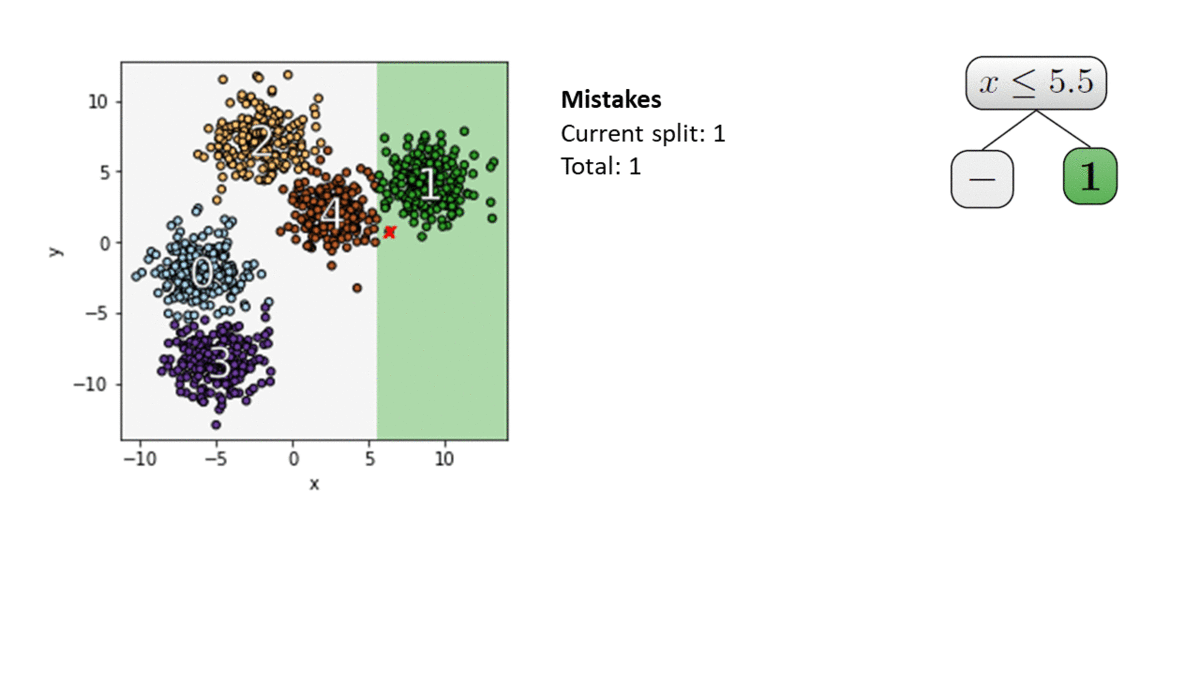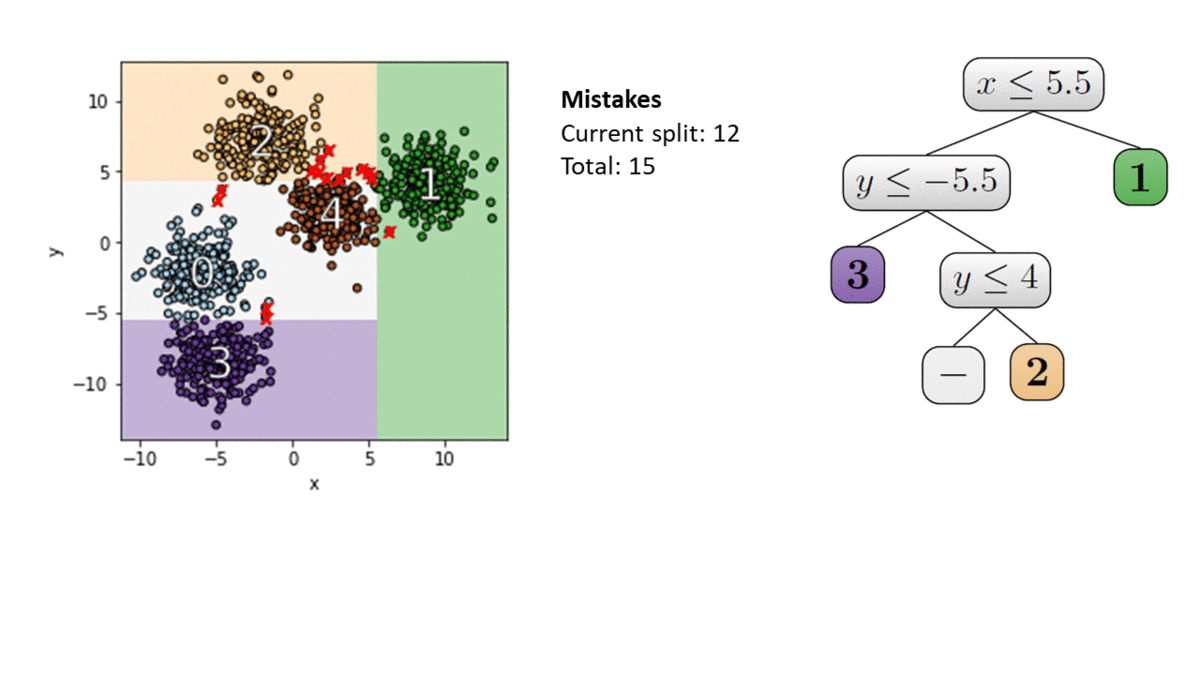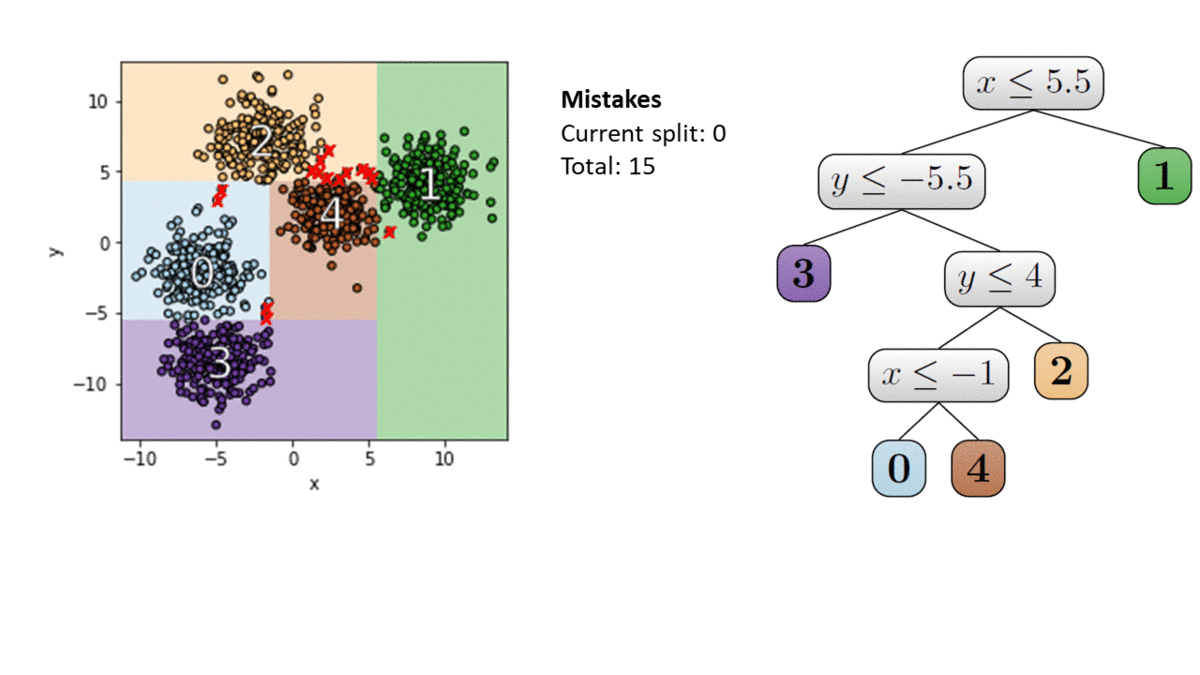
There are two important parts to FairProof : 1) How to calculate the fairness certificate (in-the-clear)? and 2) How to verify this certificate with ZKPs?
Fairness Certification in-the-clear
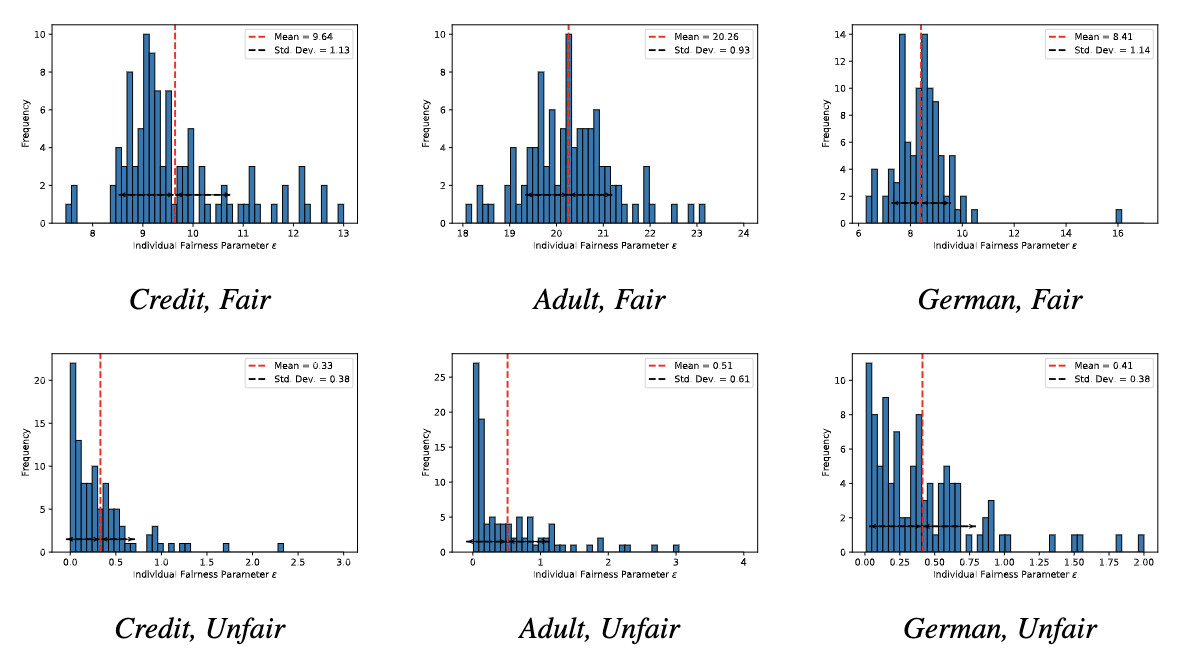
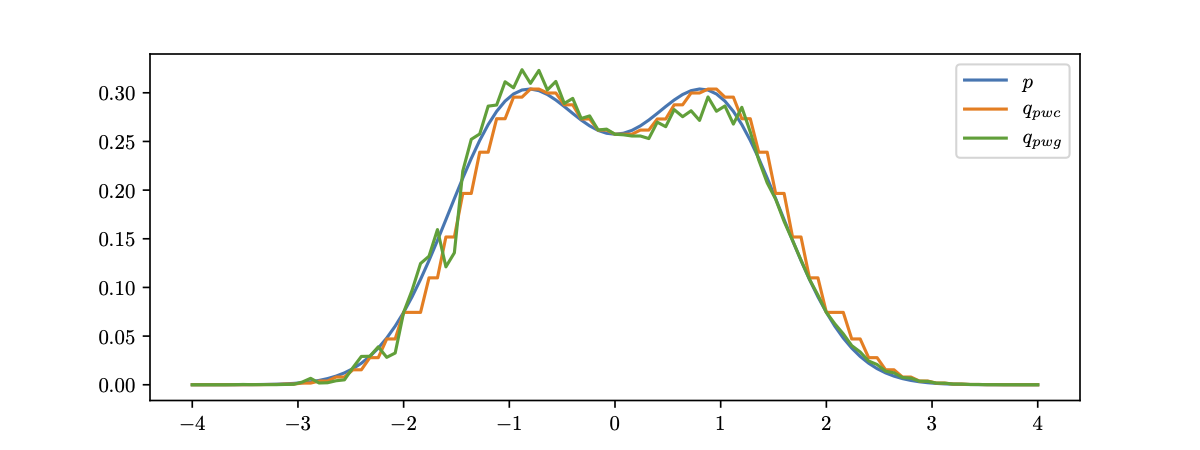
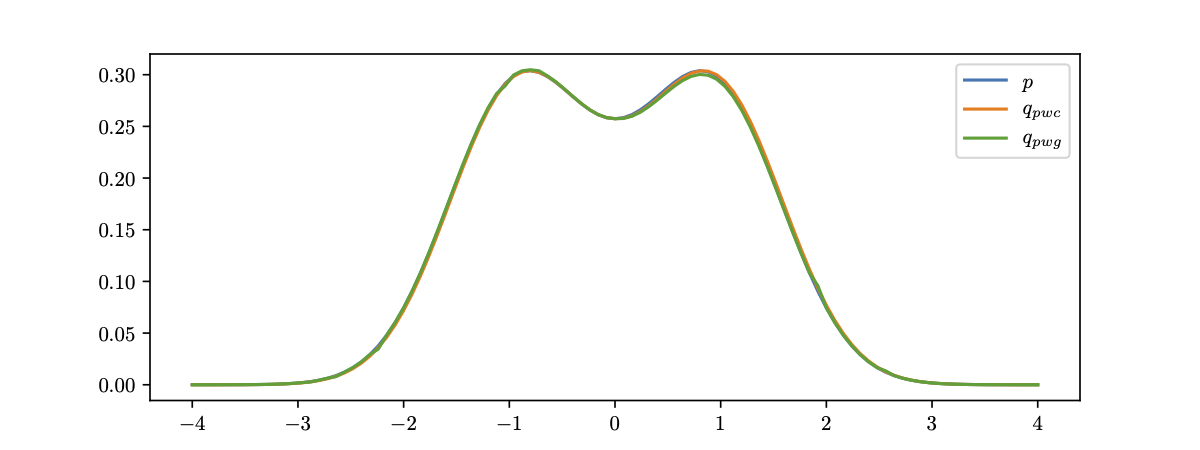
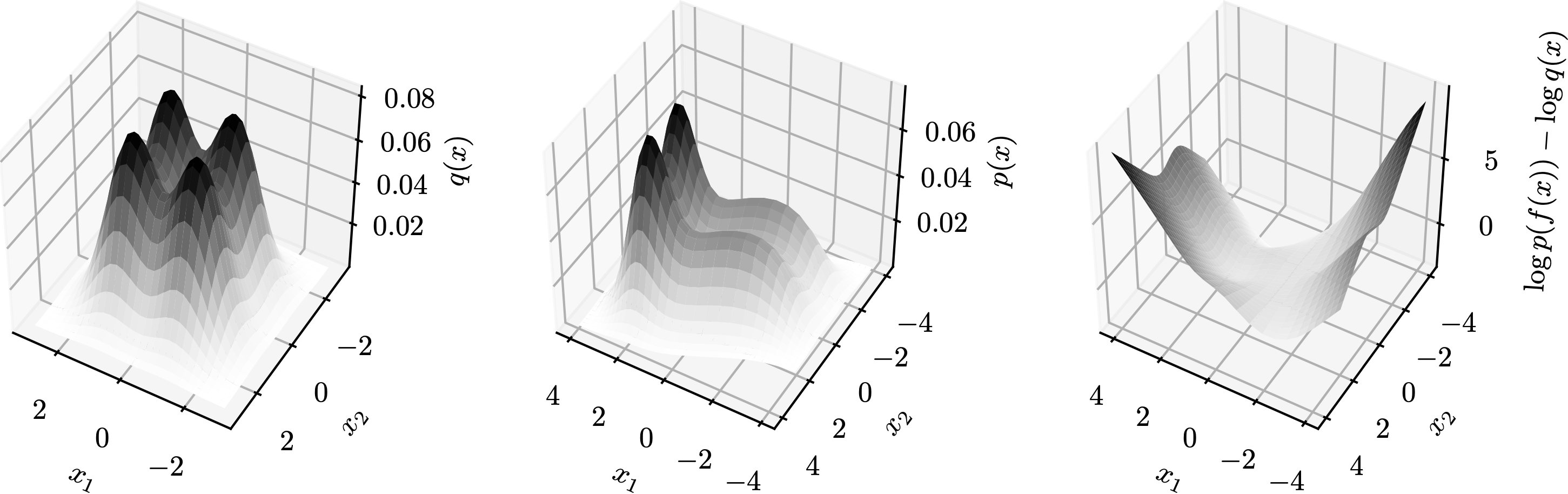
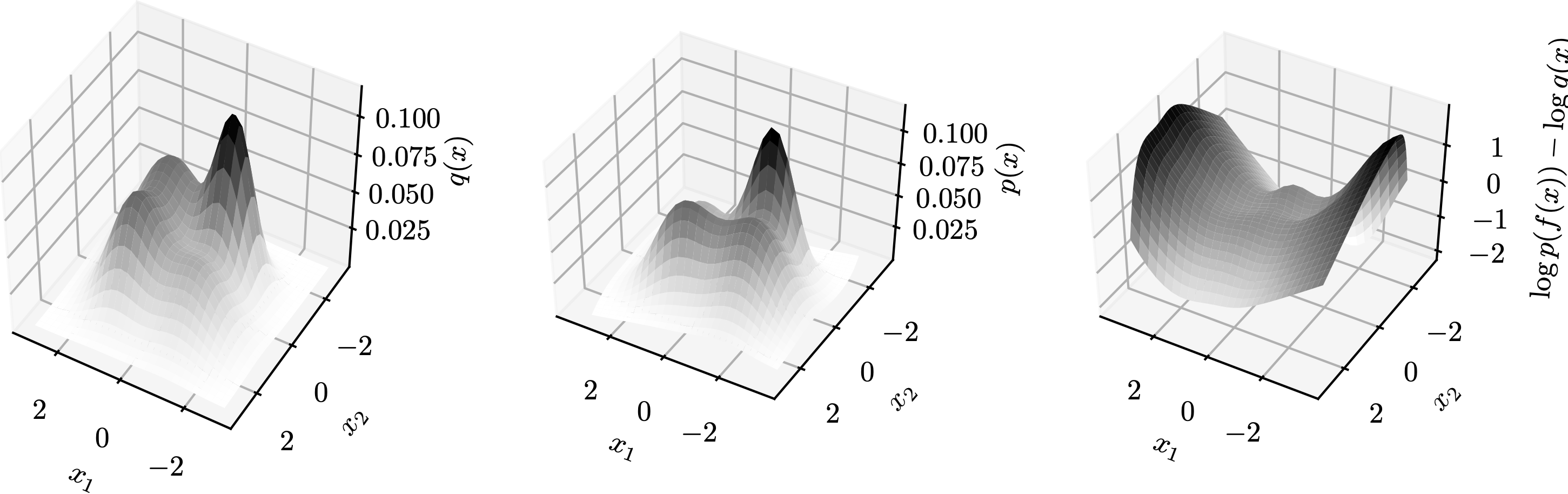
The fairness metric we use is Local Individual Fairness (IF) and give a simple algorithm to calculate this certificate by using a connection between adversarial robustness and IF. Experimentally, we see that the resulting certification algorithm is able to differentiate between less and more fair models.
Histogram of fairness parameter for fair and unfair models for 100 randomly sampled data points. Fairness parameter values are higher for more fair models.
ZKP for Fairness Certification
Next we must code this certification algorithm in a ZKP library. However, ZKPs are infamous for adding a big computational overhead and can be notoriously hard to code due to only using arithmetic operations. To overcome these challenges, we strategically choose some sub-functionalities which are enough to verify the certificate and also propose to do some computations offline to save time.
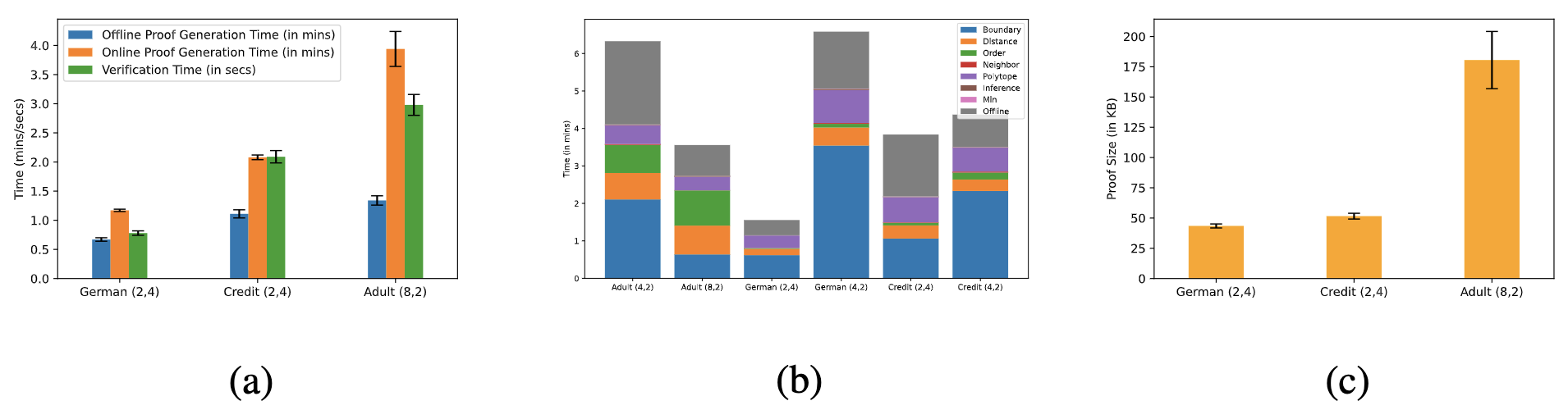
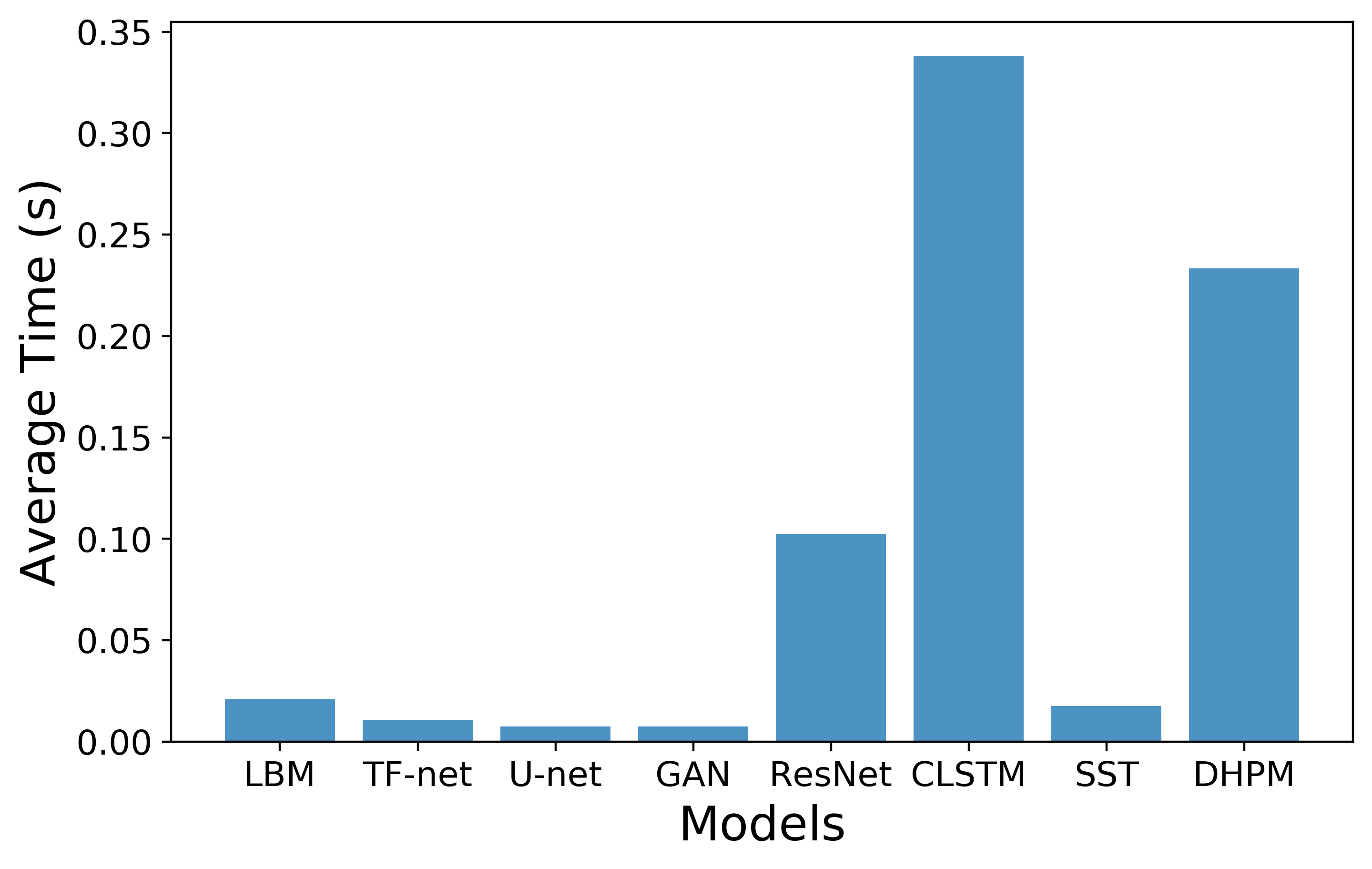
Empirically we find that the maximum proof generation time is on ~4 min while the maximum verification time is ~3 seconds (note the change from minutes to seconds). Maximum time is consumed by the VerifyNeighbor functionality. Also the proof size is a meagre 200 KB.
Results are over 100 randomly sampled points from the test sete. (a) Average Proof Generation (in mins) and Verification times (in secs) for different models. Offline computations are done in the initial setup phase while Online computations are done for every new query. Verification is only done online, for every query. (b) Breakdown of the proof generation time (in mins) for the data point with the median time. VerifyNeighbor sub-functionality takes the maximum time. (c) Average Total Proof Size (in KB) for various models. This includes the proof generated during both online and offline phases.
Conclusion
In conclusion, we propose FairProof – a protocol enabling model owners to issue publicly verifiable certificates while ensuring model confidentiality. While our work is grounded in fairness and societal applications, we believe that ZKPs are a general-purpose tool and can be a promising solution for overcoming problems arising out of the need for model confidentiality in other areas/applications as well.
]]>Chhavi YadavDYffusion: A Dynamics-informed Diffusion Model for Spatiotemporal Forecasting2023-12-22T00:00:00+00:002023-12-22T00:00:00+00:00https://ucsdml.github.io//2023/12/22/dyffusion
DYffusion forecasts a sequence of $h$ snapshots $\mathbf{x}_1, \mathbf{x}_2, \ldots, \mathbf{x}_h$
given the initial conditions $\mathbf{x}_0$ similarly to how standard diffusion models are used to sample from a distribution.
Introduction
Obtaining accurate and reliable probabilistic forecasts has a wide range of applications from
climate simulations and fluid dynamics to financial markets and epidemiology.
Often, accurate long-range probabilistic forecasts are particularly challenging to obtain .
When they exist, physics-based methods typically hinge on computationally expensive
numerical simulations .
In contrast, data-driven methods are much more efficient and have started to have real-world impact
in fields such as global weather forecasting.
Common approaches for large-scale spatiotemporal problems tend to be deterministic and autoregressive.
Thus, they are often unable to capture the inherent uncertainty in the data, produce unphysical predictions,
and are prone to error accumulation for long-range forecasts.
Diffusion models have shown great success for natural image and video generation.
However, diffusion models have been primarily designed for static data and are expensive to train and to sample from.
We study how we can efficiently leverage them for large-scale spatiotemporal problems and explicitly
incorporate the temporality of the data into the diffusion model.
Our Key Idea
We introduce a solution for these issues by designing a temporal diffusion model, DYffusion.
Following the “generalized diffusion model” framework , we
replace the forward and reverse processes of standard diffusion models
with dynamics-informed interpolation and forecasting, respectively.
This leads to a scalable generalized diffusion model for probabilistic forecasting that is naturally trained to forecast multiple timesteps.
Notation & Background
Problem setup
We study the problem of probabilistic spatiotemporal forecasting using a dataset consisting of
a time series of snapshots \(\mathbf{x}_t \in \mathcal{X}\).
We focus on the task of forecasting a sequence of \(h\) snapshots from a single initial condition.
That is, we aim to train a model to learn \(P(\mathbf{x}_{t+1:t+h} \,|\, \mathbf{x}_t)\) .
Note that during evaluation, we may evaluate the model on a larger horizon \(H>h\) by running the model autoregressively.
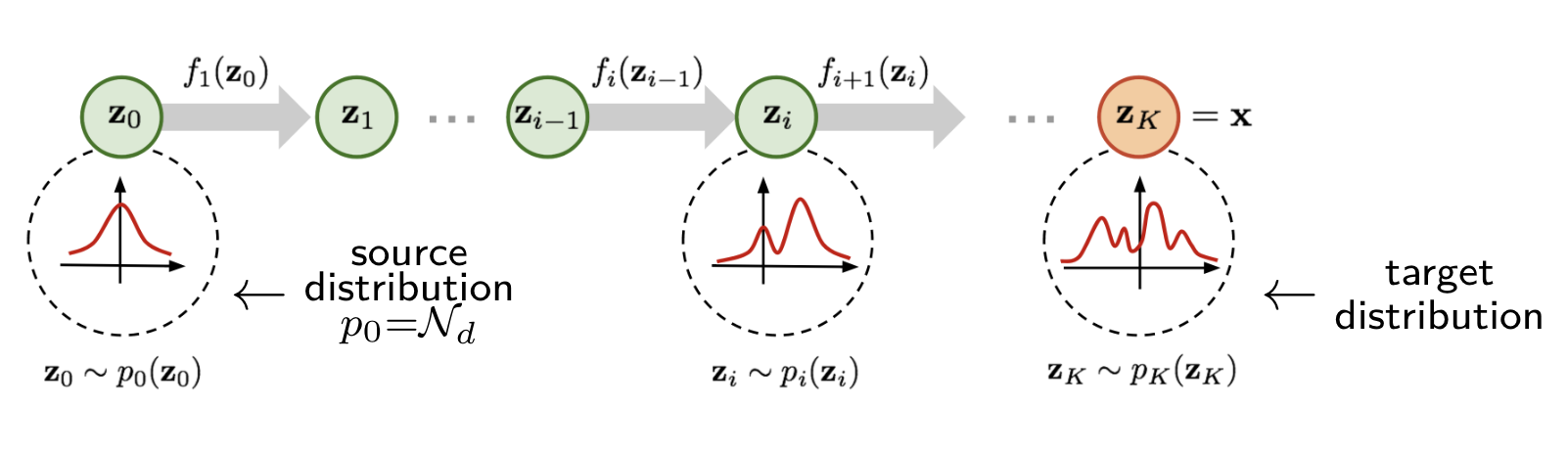
Standard diffusion models
Diffusion models iteratively transform data between an initial distribution
and the target distribution over multiple diffusion steps.
Here, we adapt the
common notation for diffusion models
to use a superscript \(n\) for the diffusion states \(\mathbf{s}^{(n)}\),
to distinguish them from the timesteps of the data, \(\mathbf{x}_t\).
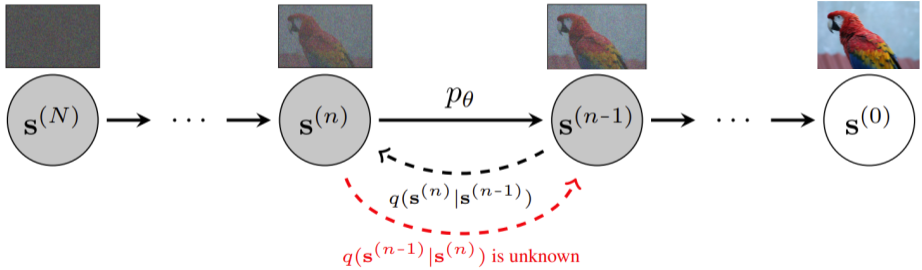
Given a data sample \(\mathbf{s}^{(0)}\), a standard diffusion model is defined through a forward diffusion process
\(q(\mathbf{s}^{(n)} \vert \mathbf{s}^{(n-1)})\)
in which small amounts of Gaussian noise are added to the sample in \(N\) steps, producing a sequence of noisy samples
\(\mathbf{s}^{(1)}, \ldots, \mathbf{s}^{(N)}\).
Adopting the notation for generalized diffusion models from , we can also consider
a forward process operator, \(D\), that outputs the corrupted samples \(\mathbf{s}^{(n)} = D(\mathbf{s}^{(0)}, n)\).
Graphical model for a standard diffusion model.
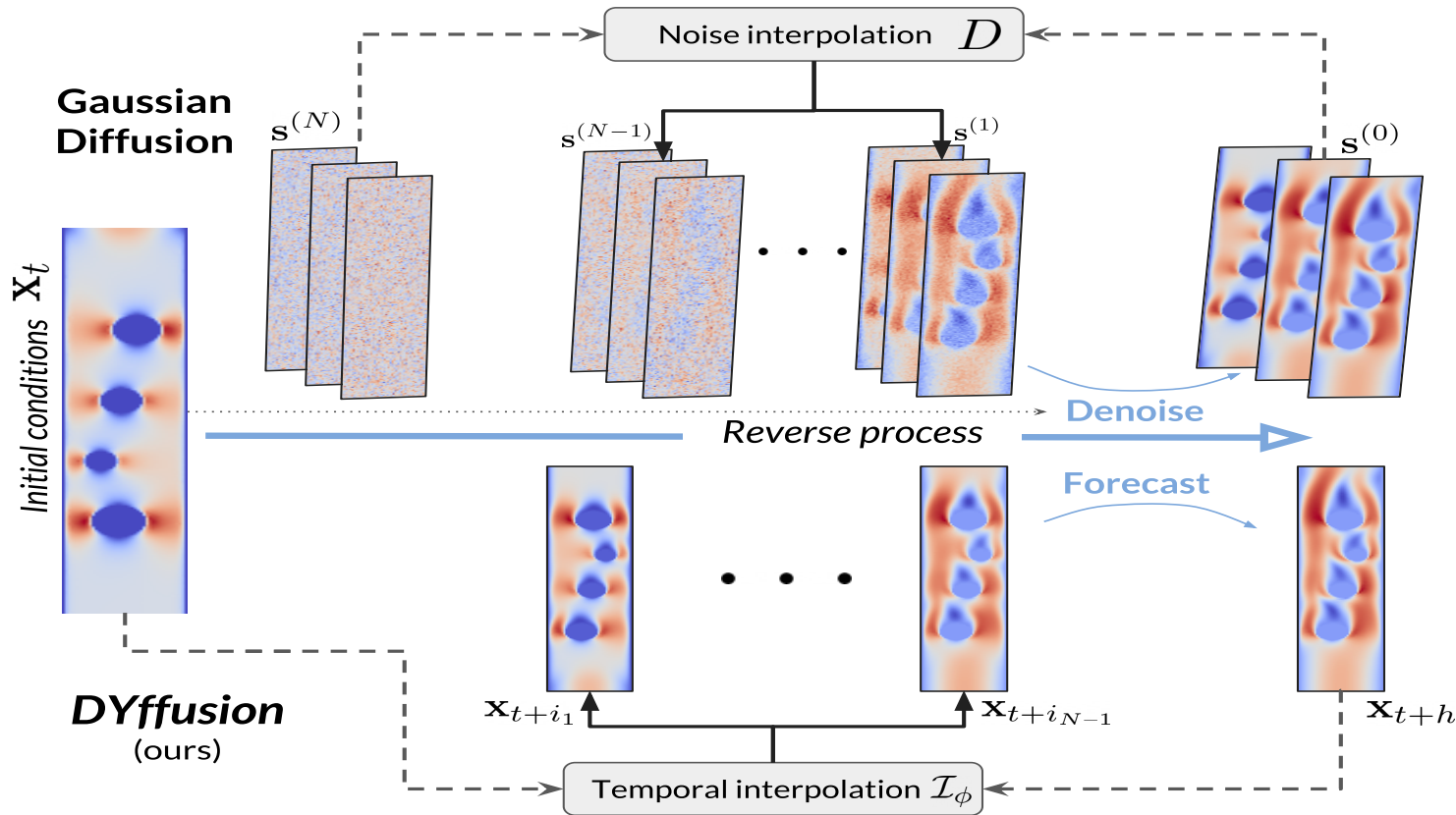
DYffusion: Dynamics-informed Diffusion Model
The key innovation of our framework, DYffusion, is a reimagining of the diffusion processes to more naturally model
spatiotemporal sequences, \(\mathbf{x}_{t:t+h}\).
Specifically, we design the reverse (forward) process to step forward (backward) in time
so that our diffusion model emulates the temporal dynamics in
the dataSimilarly to,
our forward and reverse processes cease to represent actual "diffusion" processes.
Differently to all prior work, our processes are _not_ based on data corruption or restoration..
Graphical model for DYffusion.
Implementation-wise, we replace the standard denoising network, \(R_\theta\), with a deterministic forecaster network, \(F_\theta\).
Because we do not have a closed-form expression for the forward process, we also need to learn it from data
by replacing the standard forward process operator, \(D\), with a stochastic interpolator network \(\mathcal{I}_\phi\).
Intermediate steps in DYffusion’s reverse process can be reused as forecasts for actual timesteps.
Another benefit of our approach is that the reverse process is initialized with the initial conditions of the dynamics
and operates in observation space at all times.
In contrast, a standard diffusion model is designed for unconditional generation, and reversing from white noise requires more diffusion steps.
Training DYffusion
We propose to learn the forward and reverse process in two separate stages:
Temporal interpolation as a forward process
To learn our proposed temporal forward process,
we train a time-conditioned network \(\mathcal{I}_\phi\) to interpolate between snapshots of data.
Given a horizon \(h\), we train the interpolator net so that
\(\mathcal{I}_\phi(\mathbf{x}_t, \mathbf{x}_{t+h}, i) \approx \mathbf{x}_{t+i}\) for \(i \in \{1, \ldots, h-1\}\) using the objective:
Interpolation is an easier task than forecasting, and we can use the resulting interpolator
for temporal super-resolution during inference to interpolate beyond the temporal resolution of the data.
That is, the time input can be continuous, with \(i \in (0, h-1)\).
It is crucial for the interpolator, \(\mathcal{I}_\phi\),
to produce stochastic outputs within DYffusion so that its forward process is stochastic, and it can generate probabilistic forecasts at inference time.
We enable this using Monte Carlo dropout at inference time.
Forecasting as a reverse process
In the second stage, we train a forecaster network \(F_\theta\) to forecast \(\mathbf{x}_{t+h}\)
such that \(F_\theta(\mathcal{I}_\phi(\mathbf{x}_{t}, \mathbf{x}_{t+h}, i \vert \xi), i)\approx \mathbf{x}_{t+h}\)
for \(i \in S =[i_n]_{n=0}^{N-1}\), where \(S\) denotes a schedule coupling the diffusion step to the interpolation timestep.
The interpolator network, \(\mathcal{I}\), is frozen with inference stochasticity enabled,
represented by the random variable \(\xi\).
In our experiments, \(\xi\) stands for the randomly dropped out weights of the neural network and is omitted henceforth for clarity.
Specifically, we seek to optimize the objective
To include the setting where \(F_\theta\) learns to forecast the initial conditions,
we define \(i_0 := 0\) and \(\mathcal{I}_\phi(\mathbf{x}_{t}, \cdot, i_0) := \mathbf{x}_t\).
In the simplest case, the forecaster net is supervised by all timesteps given
by the temporal resolution of the training data. That is, \(N=h\) and \(S = [j]_{j=0}^{h-1}\).
Generally, the schedule should satisfy \(0 = i_0 < i_n < i_m < h\) for \(0 < n < m \leq N-1\).
DYffusion's two-stage training procedure is summarized in the algorithm above.
Sampling from DYffusion
Our above design for the forward and reverse processes of DYffusion, implies the following generative process:
\(\begin{equation}
p_\theta(\mathbf{s}^{(n+1)} | \mathbf{s}^{(n)}, \mathbf{x}_t) =
\begin{cases}
F_\theta(\mathbf{s}^{(n)}, i_{n}) & \text{if} \ n = N-1 \\
\mathcal{I}_\phi(\mathbf{x}_t, F_\theta(\mathbf{s}^{(n)}, i_n), i_{n+1}) & \text{otherwise,}
\end{cases}
\label{eq:new-reverse}
\end{equation}\)
where \(\mathbf{s}^{(0)}=\mathbf{x}_t\) and \(\mathbf{s}^{(n)}\approx\mathbf{x}_{t+i_n}\)
correspond to the initial conditions and predictions of intermediate steps, respectively.
In our formulations, we reverse the diffusion step indexing to align with the temporal indexing of the data.
That is, \(n=0\) refers to the start of the reverse process,
while \(n=N\) refers to the final output of the reverse process with \(\mathbf{s}^{(N)}\approx\mathbf{x}_{t+h}\).
Our reverse process steps forward in time, in contrast to the mapping from noise to data in standard diffusion models.
As a result, DYffusion should require fewer diffusion steps and data.
DYffusion follows the generalized diffusion model framework.
Thus, we can use existing diffusion model sampling methods for inference.
In our experiments, we adapt the sampling algorithm from to our setting as shown below.
Sampling algorithm for DYffusion.
During the sampling process, our method essentially alternates between forecasting and interpolation,
as illustrated in the figure below.
\(R_\theta\) always predicts the last timestep, \(\mathbf{x}_{t+h}\),
but iteratively improves those forecasts as the reverse process comes closer in time to \(t+h\).
This is analogous to the iterative denoising of the “clean” data in standard diffusion models.
This motivates line 6 of Alg. 2, where the final forecast of \(\mathbf{x}_{t+h}\) can be used to
fine-tune intermediate predictions or to increase the temporal resolution of the forecast.
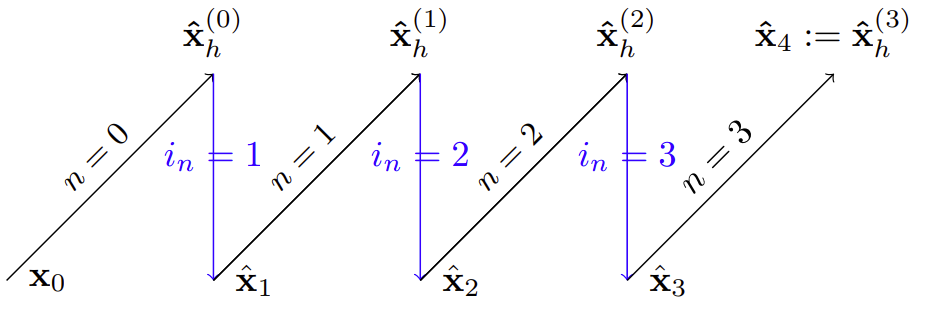
During sampling, DYffusion essentially alternates between forecasting and interpolation, following Alg. 2.
In this example, the sampling trajectory follows a simple schedule of going through all integer timesteps that precede the horizon of $h=4$,
with the number of diffusion steps $N=h$.
The output of the last diffusion step is used as the final forecast for $\hat\mathbf{x}_4$.
The black lines represent forecasts by the forecaster network, $F_\theta$.
The first forecast is based on the initial conditions, $\mathbf{x}_0$.
The blue lines represent the subsequent temporal interpolations performed by the interpolator network, $\mathcal{I}_\phi$.
Memory footprint
During training, DYffusion only requires \(\mathbf{x}_t\) and \(\mathbf{x}_{t+h}\) (plus \(\mathbf{x}_{t+i}\) during the first interpolation stage),
resulting in a constant memory footprint as a function of \(h\).
In contrast, direct multi-step prediction models including video diffusion models or (autoregressive) multi-step loss approaches require
\(\mathbf{x}_{t:t+h}\) to compute the loss.
This means that these models must fit \(h+1\) timesteps of data into memory (and may need to compute gradients recursively through them),
which scales poorly with the training horizon \(h\).
Therefore, many are limited to predicting a small number of frames or snapshots.
For example, our main video diffusion model baseline, MCVD, trains on a maximum of 5 video frames due to GPU memory constraints .
On the top row, we illustrate the direct application of a video diffusion model to dynamics forecasting for a horizon of $h=3$.
On the bottom row, DYffusion generates continuous-time probabilistic forecasts for $\mathbf{x}_{t+1:t+h}$, given the initial conditions, $\mathbf{x}_t$.
Our approach operates in the observation space at all times and does not need to model high-dimensional videos at each diffusion state.
Experimental Setup
Datasets
We evaluate our method and baselines on three different datasets:
Sea Surface Temperatures (SST): a new dataset based on NOAA OISSTv2, which
comes at a daily time-scale. Similarly to ,
we train our models on regional patches which increases the available
dataHere, we choose 11 boxes of $60$ latitude $\times 60$ longitude resolution in the eastern tropical Pacific Ocean.
Unlike the data based on the NEMO dataset in ,
we choose OISSTv2 as our SST dataset because it contains more data (although it has a lower spatial resolution of $1/4^\circ$ compared to $1/12^\circ$ of NEMO)..
We train, validate, and test all models for the years 1982-2019, 2020, and 2021, respectively.
Navier-Stokes flow benchmark dataset from , which consists of a
\(221\times42\) grid. Each trajectory contains four randomly generated circular obstacles that block the flow.
The channels consist of the \(x\) and \(y\) velocities as well as a pressure field and the viscosity is \(1e\text{-}3\).
Boundary conditions and obstacle masks are given as additional inputs to all models.
Spring Mesh benchmark dataset from . It represents a \(10\times10\) grid of
particles connected by springs, each with mass 1. The channels consist of two position and momentum fields each.
We follow the official train, validation, and test splits from for the Navier-Stokes and spring mesh datasets,
always using the full training set for training.
Baselines
We compare our method against both direct applications of standard diffusion models to dynamics forecasting and
methods to ensemble the “barebone” backbone network of each dataset. The network operating in “barebone” form means
that there is no involvement of diffusion.
We use the following baselines:
DDPM: We train it as a multi-step (video-like problem) conditional diffusion model.
MCVD: A state-of-the-art conditional video diffusion modelWe train MCVD in "concat" mode, which in their experiments performed best..
Dropout: Ensemble multi-step forecasting of the barebone backbone network based on enabling dropout at inference time.
Perturbation: Ensemble multi-step forecasting with the barebone backbone network based on random perturbations of the initial conditions with a fixed variance.
Official deterministic baselines from for
the Navier-Stokes and spring mesh datasets Due to their deterministic nature, we exclude these baselines from our main probabilistic benchmarks..
MCVD and the multi-step DDPM predict the timesteps \(\mathbf{x}_{t+1:t+h}\) based on \(\mathbf{x}_{t}\).
The barebone backbone network baselines are time-conditioned forecasters trained on the multi-step objective
\(\mathbb{E}_{i \sim \mathcal{U}[\![1, h]\!], \mathbf{x}_{t, t+i}\sim \mathcal{X}}
\| F_\theta(\mathbf{x}_{t}, i) - \mathbf{x}_{t+i}\|^2\)
from scratchWe found it to perform very similarly to predicting all $h$
horizon timesteps at once in a single forward pass, i.e. on the
objective $\mathbb{E}_{\mathbf{x}_{t:t+h}\sim \mathcal{X}} \| F_\theta(\mathbf{x}_{t}) - \mathbf{x}_{t+1:t+h}\|^2$.
Neural network architectures
For a given dataset, we use the same backbone architecture for all baselines as well as for both the interpolation and forecaster networks in DYffusion.
For the SST dataset, we use a popular UNet architecture designed for diffusion models.
For the Navier-Stokes and spring mesh datasets, we use the UNet and CNN from the original benchmark paper , respectively.
The UNet and CNN models from are extended by the sine/cosine-based featurization module of the SST UNet to embed the diffusion step or dynamical timestep.
Evaluation metrics
We evaluate the models by generating an M-member ensemble (i.e. M samples are drawn per batch element), where
we use M=20 for validation and M=50 for testing.
As metrics, we use the Continuous Ranked Probability Score (CRPS) ,
the mean squared error (MSE), and the spread-skill ratio (SSR).
The CRPS is a proper scoring rule and a popular metric in the probabilistic forecasting
literature.
The MSE is computed on the ensemble mean prediction.
The SSR is defined as the ratio of the square root of the ensemble variance to the corresponding ensemble mean RMSE.
It serves as a measure of the reliability of the ensemble, where values smaller than 1 indicate
underdispersionThat is, the probabilistic forecast is overconfident and fails to model the full uncertainty of the forecast
and larger values overdispersion.
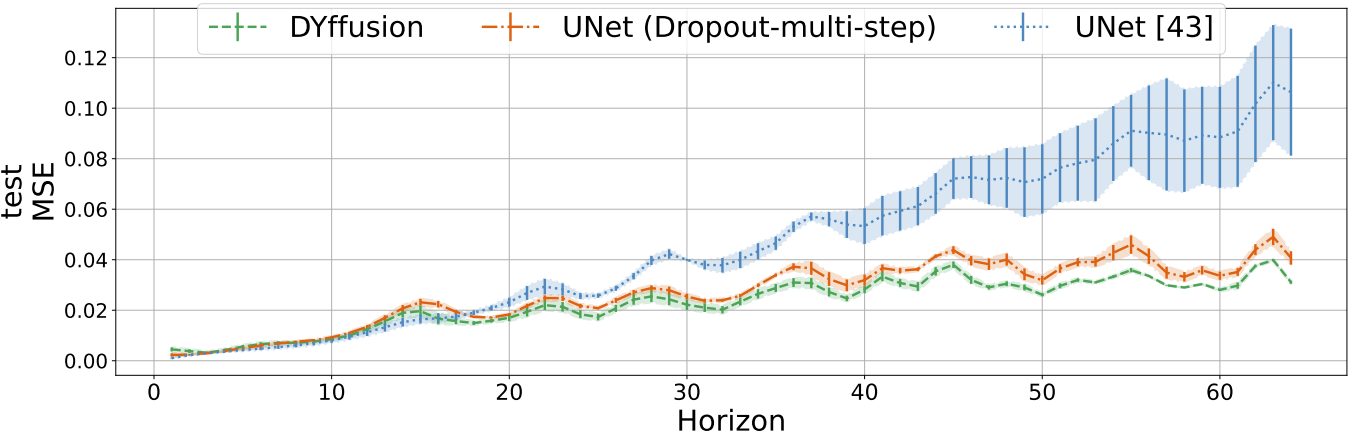
On the Navier-Stokes and spring mesh datasets, models are evaluated by autogressively forecasting the full test trajectories of length 64 and 804, respectively.
For the SST dataset, all models are evaluated on forecasts of up to 7 daysWe do not explore more long-term SST forecasts because the chaotic nature of the system, and the fact that we only use regional patches, inherently limits predictability..
Results
Quantitative results
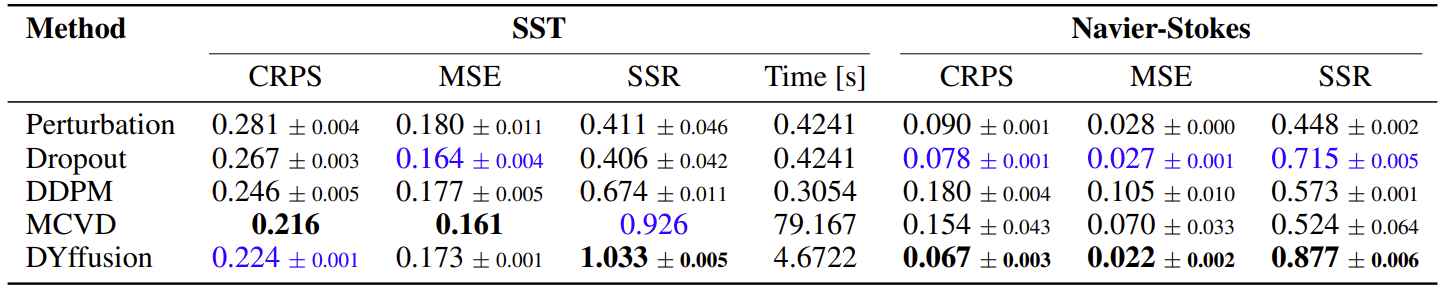
We present the time-averaged metrics for the SST and Navier-Stokes dataset in the table below.
DYffusion performs best on the Navier-Stokes dataset, while coming in a close second on the SST dataset after MCVD, in terms of CRPS.
Since MCVD uses 1000 diffusion steps,
it is slower to sample from at inference time than from DYffusion, which is trained with at most 35 diffusion steps.
The DDPM model for the SST dataset is fairly efficient because it only uses 5 diffusion steps but lags in terms of performance.
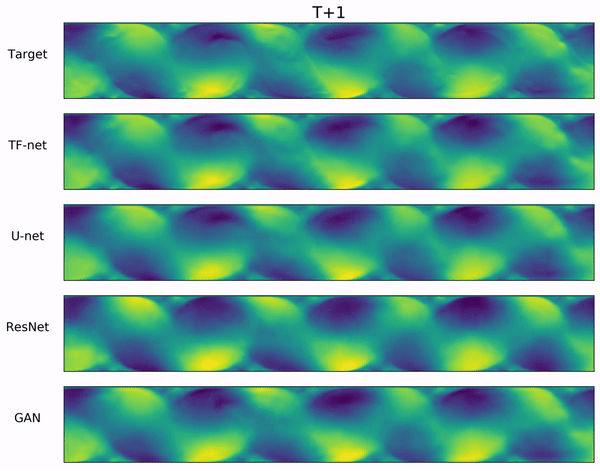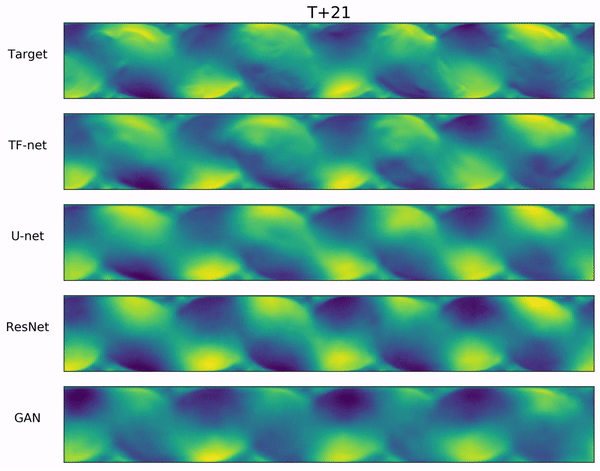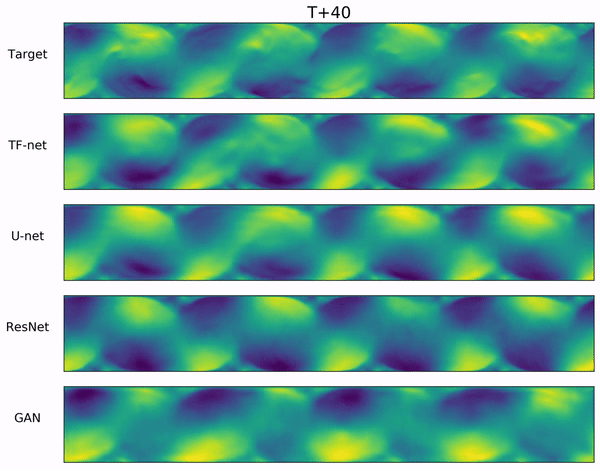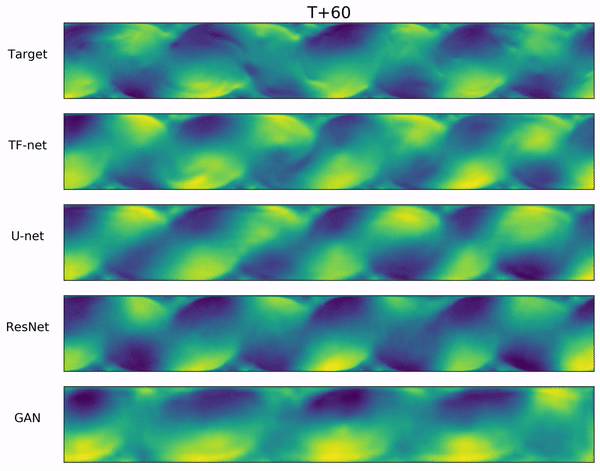
Results for sea surface temperature forecasting of 1 to 7 days ahead, and Navier-Stokes
flow full trajectory forecasting of 64 timesteps.
For SST, all models are trained on forecasting $h=7$ timesteps. The time column represents the time needed to forecast all 7 timesteps for a single batch.
For Navier-Stokes, Perturbation, Dropout, and DYffusion are trained on a horizon of $h=16$.
MCVD and DDPM are trained on $h=4$ and $h=1$, respectively, as we could not successfully train them using larger horizons.
Bold indicates best, blue second best.
For CRPS and MSE, lower is better. For SSR, closer to 1 is better. Numbers are averaged out over the evaluation horizon.
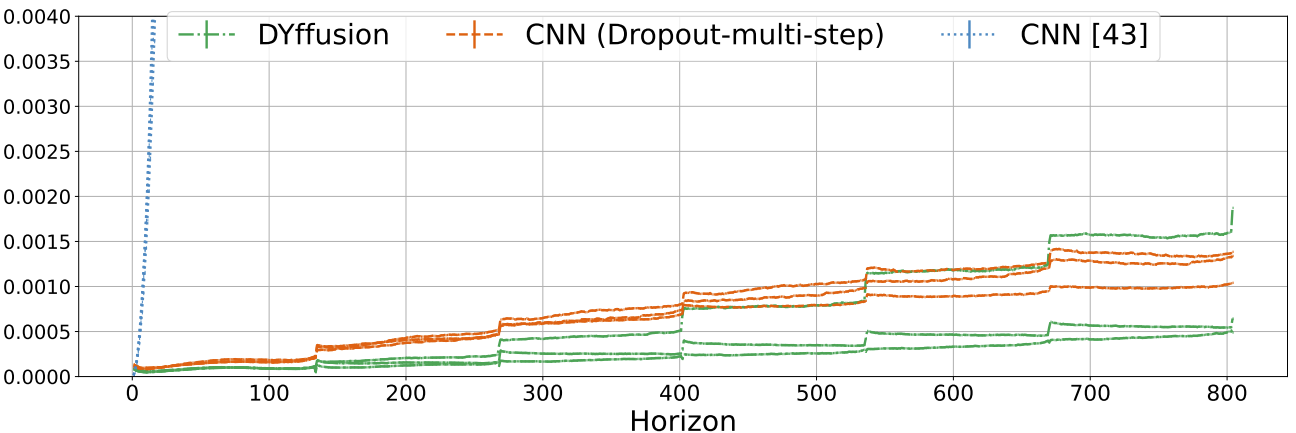
Thanks to the dynamics-informed and memory-efficient nature of DYffusion, we can scale our framework to long horizons.
On the spring mesh dataset, we train with a horizon of 134 and evaluate the models on trajectories of 804 time steps.
Our method beats the Dropout baseline, with a larger margin on the out-of-distribution test dataset.
Despite several attempts with varying hyperparameter configurations neither the DDPM nor the MCVD diffusion model converged on this dataset.
Spring Mesh results. Both methods are trained on a horizon of $h = 134$ timesteps and
evaluated how well they forecast the full test trajectories of 804 steps.
For CRPS and MSE, lower is better. For SSR, closer to 1 is better. Numbers are averaged out over the evaluation horizon.
The reported MSE scores above, using the same CNN architecture,
are significantly better than the ones reported for the official CNN baselines in Fig. 8 of ,
where the deterministic CNN diverged or attained a very poor MSE.
This is likely because our models are trained to forecast multiple timesteps,
while the models from are trained to forecast the next timestep only.
As a result, the training objective significantly deviates from the evaluation procedure,
which was already noted as a limitation of the benchmark baselines in .
This effect is also found for the Navier-Stokes dataset to a lower extent, as demonstrated in the figures below.
Navier-Stokes
Spring Mesh
Comparison against single-step deterministic baselines from .
We plot the MSE as a function of the rollout time step.
For spring mesh, we plot each of the three models trained with a different random seed separately due to the high variance.
Qualitative results
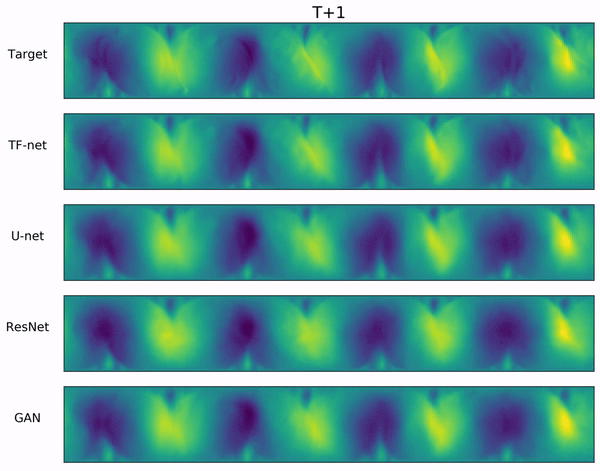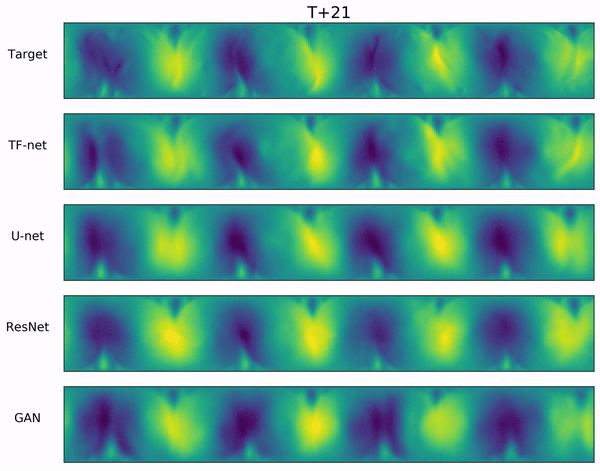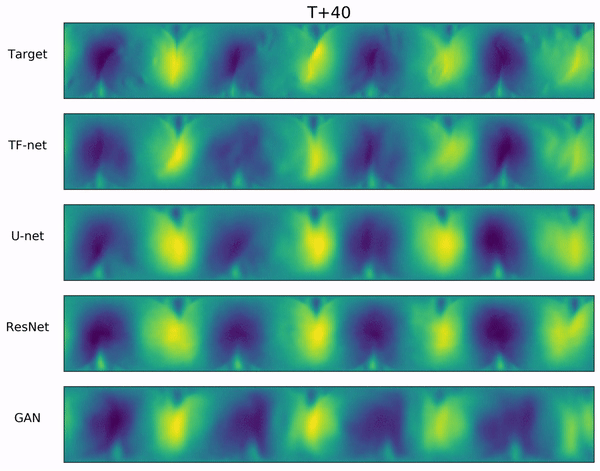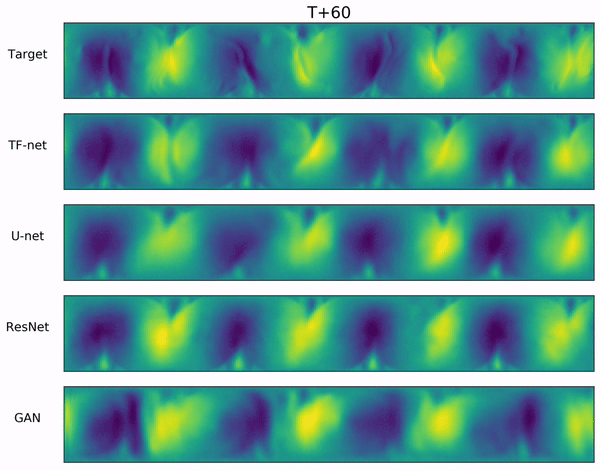
Long-range forecasts of ML models often suffer from blurriness or might even diverge when using autoregressive models.
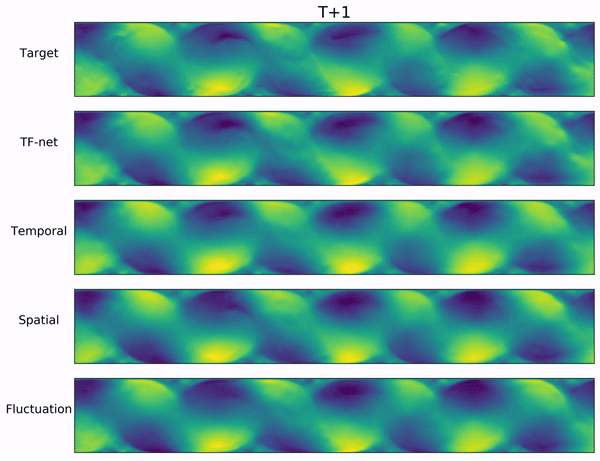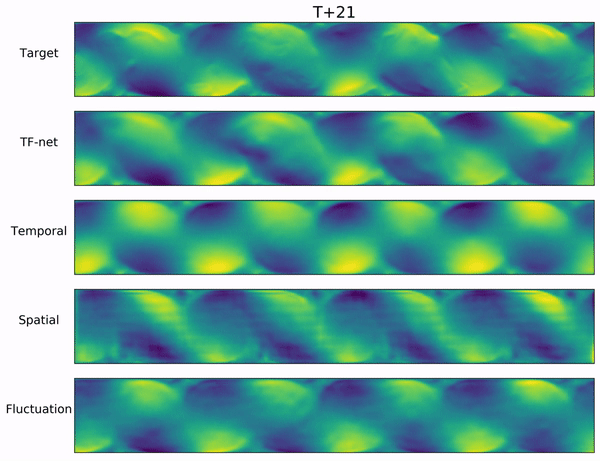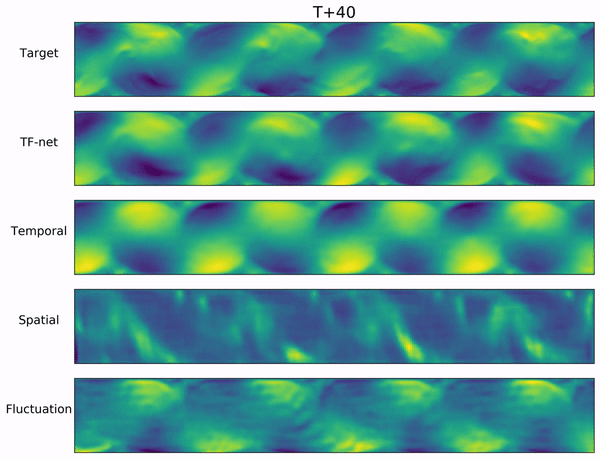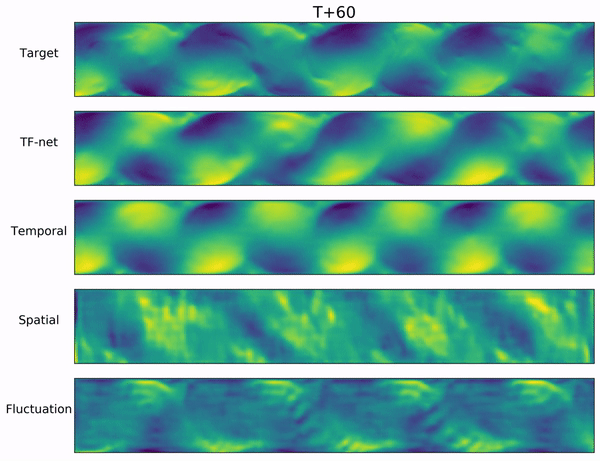
In the video below, we show a complete Navier-Stokes test trajectory forecasted by DYffusion and the best baseline, Dropout, as well as the corresponding ground truth.
Our method can reproduce the true dynamics over the full trajectory and does so better than the baseline,
especially for fine-scale patterns such as the tails of the flow after the right-most obstacle.
Exemplary samples from DYffusion and the best baseline, Dropout, as well as the corresponding ground truth from a complete Navier-Stokes trajectory forecast.
Temporal super-resolution and sample variability
Motivated by the continuous-time nature of DYffusion, we aim to study in this experiment whether it is possible to forecast
skillfully beyond the resolution given by the data.
Here, we forecast the same Navier-Stokes trajectory shown in the video above but at \(8\times\) resolution.
That is, DYffusion forecasts 512 timesteps instead of 64 in total.
This behavior can be achieved by either changing the sampling trajectory \([i_n]_{n=0}^{N-1}\) or
by including additional output timesteps, \(J\), for the refinement step of line 6 in Alg. 2.
In the video below, we choose to do the latter and find the 5 sampled forecasts to be visibly pleasing and temporally consistent with the ground truth.
$8\times$ temporal super-resolution of a Navier-Stokes trajectory with DYffusion.
The ground truth is frozen in-between the original timesteps. Five distinct samples are shown.
Note that we hope that our probabilistic forecasting model can capture any of the possible,
uncertain futures instead of forecasting their mean, as a deterministic model would do.
As a result, some long-term rollout samples are expected to deviate from the ground truth.
For example, see the velocity at t=3.70 in the video above.
It is reassuring that DYffusion’s samples show sufficient variation, but also cover the ground truth quite well (sample 1).
This advantage is also reflected quantitatively in the spread-skill ratio (SSR) metric, where DYffusion
consistently reached values close to 1.
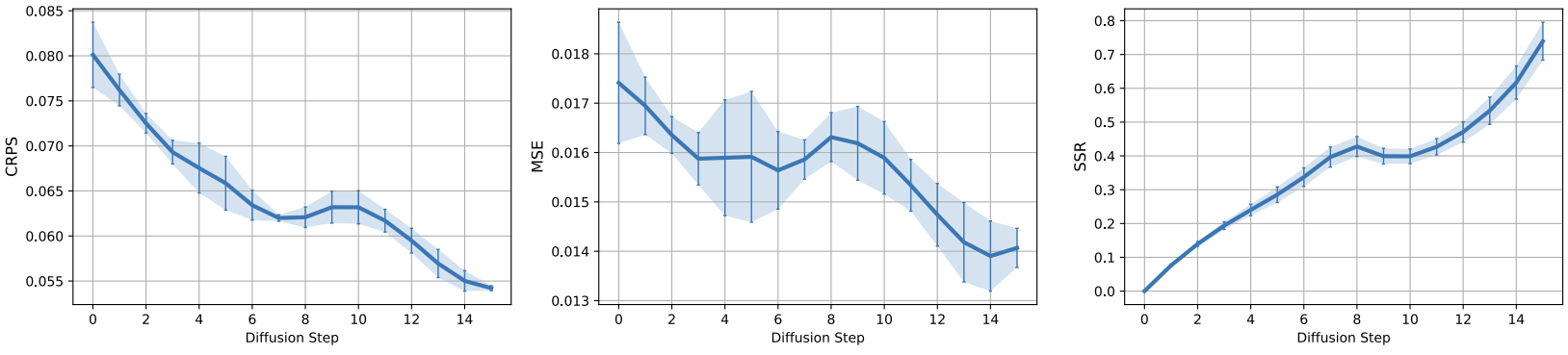
Iterative refinement of forecasts
DYffusion’s forecaster network repeatedly predicts the same timestep, \(t+h\), during sampling.
Thus, we need to verify that these forecasts,
\(\hat{\mathbf{x}}_{t+h} = F_\theta(\mathbf{x}_{t+i_n}, i_n)\), tend to improve throughout the course of the reverse process,
i.e. as \(n\rightarrow N\) and \(\mathbf{x}_{t+i_n}\rightarrow\mathbf{x}_{t+h}\).
Below we show that this is indeed the case for the Navier-Stokes dataset.
Generally, we find that this observation tends to hold especially for the probabilistic metrics, CRPS and SSR,
while the trend is less clear for the MSE across all datasets (see Fig. 7 of our paper).
DYffusion's forecaster network iteratively improves its forecasts during sampling.
Conclusion
DYffusion is the first diffusion model that relies on task-informed forward and reverse processes.
Other existing diffusion models, albeit more general, use data corruption-based processes.
Thus, our work provides a new perspective on designing a capable diffusion model,
and we hope that it will lead to a whole family of task-informed diffusion models.
If you have any application that you think could benefit from DYffusion, or build on top of it, we would love to hear from you!
]]>Salva Rühling CachayHow to Actively Learn in Bounded Memory2021-11-21T17:00:00+00:002021-11-21T17:00:00+00:00https://ucsdml.github.io//jekyll/update/2021/11/21/al-memoryA Brief Introduction: Enriched Queries and Memory Constraints
In the world of big-data, machine learning practice is dominated by massive supervised algorithms, techniques that require huge troves of labeled data to reach state of the art accuracy. While certainly successful in their own right, these methods break down in important scenarios like disease classification where labeling is expensive, and accuracy can be the difference between life and death. In a previous post, we discussed a new technique for tackling these high risk scenarios using enriched queries: informative questions beyond labels (e.g., comparing data points). While the resulting algorithms use very few labeled data points and never make errors, their efficiency comes at a cost: memory usage.
For simplicity, in this post we’ll consider the following basic setup. Let $X$ be a set of $n$ labeled points, where the labeling is chosen from some underlying family of classifiers (e.g., linear classifiers). As the learner, we are given access to the (unlabeled) points in $X$, a labeling oracle we can call to learn the label of any particular $x \in X$, and a set of special enriched oracles that give further information about the underlying classifier (e.g., a comparison oracle which can compare any two points $x,x’ \in X$). Our goal is to learn the label of every point in $X$ in as few queries (calls to the oracle) as possible.
Traditional techniques for solving this problem aim to use only $\log(n)$ adaptive queries. For instance if $X$ is a set of points on the real line and the labeling is promised to come from some threshold, we can achieve this using just a labeling oracle and binary search. This gives an exponential improvement over the naive algorithm of requesting the label of every point! However, these strategies generally have a problem: in order to choose the most informative queries, they allow the algorithm access to all of $X$, implicitly assuming the entire dataset is stored in memory. Since we frequently deal with massive datasets in practice, this strategy quickly becomes intractable. In this post, we’ll discuss a new compression-based characterization of when its possible to learn in $\log(n)$ queries, but store only a constant number of points in the process.
A Basic Example: Learning Thresholds via Compression
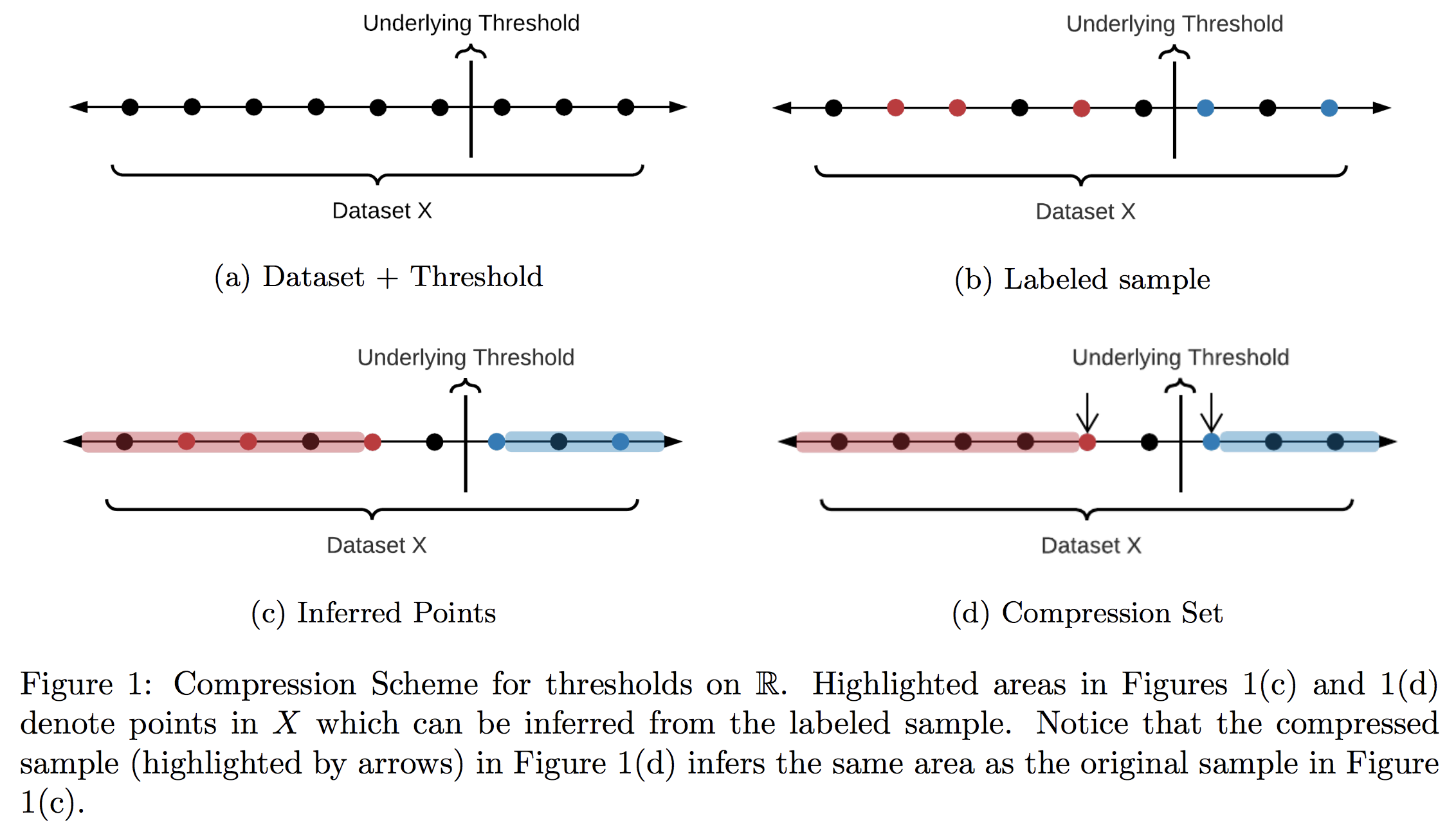
Learning in constant memory may seem a tall order when the algorithm is already required to correctly recover every label in a size $n$ set $X$ in only $\log(n)$ queries. To convince the reader such a feat is even possible, let’s start with a fundamental example using only label queries: thresholds in 1D. Let $X$ be any set of $n$ points on $\mathbb{R}$ with (hidden) labels given by some threshold. We’d like to learn the label of every point in $X$ in around $\log(n)$ adaptive queries of the form “what is the label of $x \in X$?” Notice that to do this, it is enough to find the points directly to the right and left of the threshold—the only issue is we don’t know where they are! Classically, we’d try find these points using binary search. This would acheive the $\log(n)$ bound on queries, but determining which point to query in each step requires too much memory.
A better strategy for this problem was proposed by Kane, Lovett, Moran, and Zhang (KLMZ). They follow a simple four step process:
Randomly sample $O(1)$ points from remaining set (initially $X$ itself).
Query the labels of these points, and store them in memory.
Restrict to the set of points whose labels remain unknown.
Repeat $O(\log(n))$ times.
Note that it is possible to remove points we have not queried in Step 3 (we call such points “inferred,” see Figure 1(c)). Indeed, KLMZ prove that despite only making $O(1)$ queries, each round should remove about half of the remaining points. As a result, after about $\log(n)$ rounds, we must have found the two points on either side of the threshold, and can therefore label all of $X$ as desired (see our previous post for more details on this algorithm). This algorithm is much better than binary search, but it still stores $O(\log(n))$ points overall—we’d like an algorithm whose memory doesn’t scale with $n$ at all!
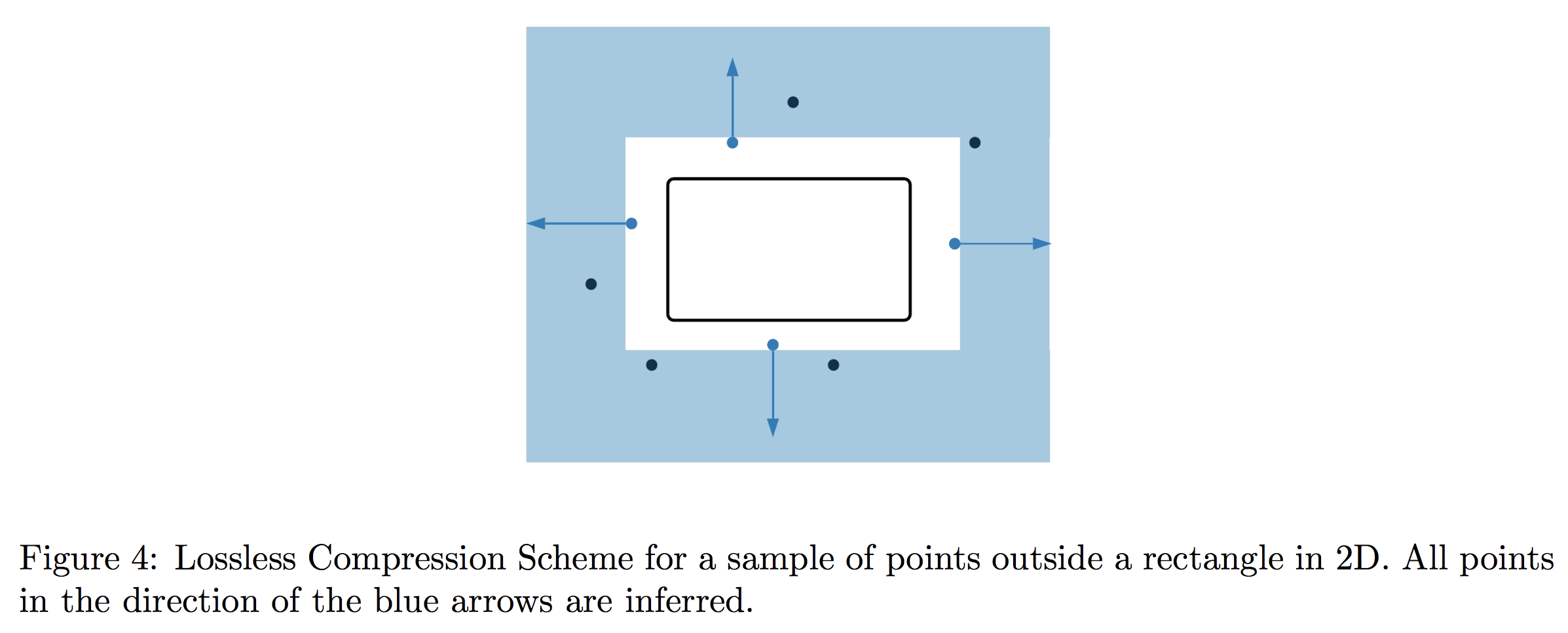
It turns out that for the class of thresholds, this can be achieved by a very simple tactic: in each round, only store the two points closest to each side of the threshold. This “compressed” version of the sample actually retains all relevant information, so the algorithm’s learning guarantees are completely unaffected. Let’s take a look pictorially.
Since we can compress our storage down to a constant size in every round and never draw more than $O(1)$ points, this strategy results in a learner whose memory has no dependence on $X$ at all: a zero-error, query efficient, bounded memory learner.
A General Framework: Lossless Sample Compression
Our example for thresholds in 1D suggests the following paradigm: if we can compress samples down to $O(1)$ points without harming inference, bounded memory learning is possible. This is true, but not particularly useful: most classes beyond thresholds can’t even be actively learned (e.g., halfspaces in $2D$), much less in bounded memory. To build learners for classes beyond thresholds, we’ll need to generalize our idea of compression to the enriched query regime. In more detail, let $X$ be a set and $H$ a family of binary labelings of $X$. We consider classes $(X,H)$ with an additional query set $Q$. Formally, $Q$ consists of a set of oracles that contain information about the set $X$ based upon the structure of the underlying hypothesis $h \in H$. Our formal definition of these oracles is fairly broad (see our paper for exact details), but they can be thought of simply as functions dependent on the underlying hypothesis $h \in H$ that give additional structural information about tuples in $X$. One standard example is the comparison oracle on halfspaces. Given a particular halfspace $\langle \cdot, v \rangle$, the learner may send a pair $x,x’$ to the comparison oracle to learn which example is closer to the decision boundary, or equivalently they recieve $\text{sign}(\langle x, v \rangle - \langle x’, v \rangle)$).
To generalize our compression-based strategy for thresholds to the enriched query setting, we also need to discuss a little bit of background on the theory of inference. Let $(X,H)$ be a hypothesis class with associated query set $Q$. Given a sample $S \subset X$ and query response $Q(S)$, denote by $H_{Q(S)}$ the set of hypotheses consistent with $Q(S)$ (also called the version space, this is the set of $h \in H$ such that $Q(S)$ is a valid response if $h$ is the true underlying classifier). We say that $Q(S)$ infers some $x \in X$ if all consistent classifiers label $x$ the same, that is if there exists $z \in$ {$0,1$} such that:
\[
\forall h \in H_{Q(S)}, h(x)=z.
\]
This allows us to label $x$ with 100% certainty, since the true underlying classifier must lie in $H_{Q(S)}$ by definition, and all such classifiers give the same label to $x$!
In the case of thresholds, our compression strategy relied on the fact that the two points closest to the boundary inferred the same amount of information as the original sample. We can extend this idea naturally to the enriched query regime as well.
Let $X$ be a set and $H$ a family of binary classifiers on $X$. We say $(X,H)$ has a lossless compression scheme (LCS) $W$ of size $k$ with respect to a set of enriched queries $Q$ if for all subsets $S \subset X$ and all query responses $Q(S)$, there exists a subset $W = W(Q(S)) \subseteq S$ such that $|W| \leq k$, and any point in $X$ whose label is inferred by $Q(S)$ is also inferred by $Q(W)$.
Recall our goal is to correctly label every point in $X$. Using lossless compression, we can now state our general algorithm for this process:
Randomly sample $O(1)$ points from remaining set (initially $X$ itself).
Make all queries on these points, and store them in memory.
Compress memory via the lossless compression scheme.
Restrict to the set of points whose labels remain unknown.
Before moving on to some examples, let’s take a brief moment to discuss the proof. The result essentially follows in two steps. First, we’d like to show that for any distribution over $X$, drawing $O(k)$ points is sufficient to infer $1/2$ of $X$ in expectation. This follows similarly to standard results in the literature—one can either use the classic sample compression arguments of Floyd and Warmuth, or more recent symmetry arguments of KLMZ. With this in hand, it’s easy to see that after $\log(n)$ rounds (learning $1/2$ of $X$ each round), we’ll have learned all of $X$. The second step is then to observe that our compression in each step has no effect on this learning procedure. This follows without too much difficulty from the definition of lossless sample compression, which promises that the compressed sub-sample preserves all such information.
Example: Axis-Aligned Rectangles
While interesting in its own right, a sufficient condition like Lossless Sample Compression is most useful if it applies to natural classifiers. We’ll finish our post by discussing an application of this paradigm to labeling a dataset $X$ when the underlying classifier is given by an axis-aligned rectangle. Axis-aligned Rectangles are a natural generalization of intervals to higher dimensions. They are given by a product of $d$ intervals in $\mathbb{R}$:
\[
R = \prod\limits_{i=1}^d [a_i,b_i],
\]
such that an example $x=(x_1,\ldots,x_d) \in \mathbb{R}^d$ lies in the rectangle if every feature lies inside the specified interval, that is $x_i \in [a_i,b_i]$.
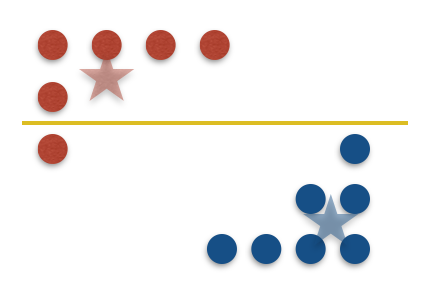
Standard arguments show that with only labels, learning the labels of a set $X$ of size $n$ takes $\Omega(n)$ queries in the worst case when the labeling is given by some underlying rectangle. To see why, let’s consider the simple case of 1D—intervals. The key observation is that a sample of points $S_{\text{out}}$ lying outside the interval cannot infer any information beyond its own labels. This is because for any $x \in \mathbb{R} \setminus S_{\text{out}}$, there exists an interval that includes $x$ but not $S_{\text{out}}$ (say $I=[x-\varepsilon,x+\varepsilon]$ for some small enough $\varepsilon$), and an interval that excludes $x$ and $S_{\text{out}}$ (say $I=[x+\varepsilon,x+2\varepsilon]$). As a result, we cannot tell whether $x$ is included in the underlying interval. In turn, this means that if we try to compress $S_{\text{out}}$ in any way, we will always lose information about the original sample.
To circumvent this issue, we introduce “odd-one-out” queries. This new query type allows the learner to take any point $x\in X$ in the dataset that lies outside of the rectangle $R$, and ask for a violated coordinate (i.e. a feature lying outside one of the specified intervals) and the direction of violation (was the coordinate too large, or too small?). Concretely, imagine a chef is trying to cook a dish for a particularly picky patron. After each failed attempt, the chef asks the patron what went wrong, and the patron responds with some feature they dislike (perhaps the meat was overcooked, or undersalted). It turns out that such scenarios have small lossless compression schemes (and are therefore learnable in bounded memory).
Axis-Aligned Rectangles over $\mathbb{R}^d$ have an $O(d)$-size LCS with respect to label and odd-one-out queries.
We’ll wrap up our post by sketching the proof. It will be convenient to break our compression scheme into two parts: a scheme for points inside the rectangle, and a scheme points outside the rectangle.1
Let’s start with the former case and restrict our attention to a sample of points $S_{\text{in}}$ that lies entirely inside the rectangle. We claim that all the relevant information in this case is captured by the maximum and minimum values of coordinates in $S_{\text{in}}$. Storing the $2d$ points achieving these values can be viewed as storing a bounding box that is guaranteed to lie inside the underlying rectangle classifier.
Notice that for any point $x \in \mathbb{R}^d$ outside of the bounding box, the version space (that is the set of all rectangles that contain $S_{\text{in}}$) has both a rectangle that contains $x$, and a rectangle that does not contain $x$. This means that label queries on $S_{\text{in}}$ cannot infer any point outside of the bounding box. Since every point inside the box is inferred by the compressed sample, these $2d$ points give a compression set for $S_{\text{in}}$.
Now let’s restrict our attention to a sample $S_{\text{out}}$ that lies entirely outside the rectangle. In this case, we’ll additionally have to compress information given by the odd-one-out oracle as well as labels. Nevertheless, we claim that a simple strategy suffices: store the closest point to each edge of the rectangle.
In particular, because the odd-one-out oracle gives a violated coordinate and direction of violation, any point that is further out in the direction of violation must also lie outside the rectangle. In any given direction, it is not hard to see that all relevant information is captured by the closest point to the relevant edge, since any further point can be inferred to be too far in that direction.
Conclusion
We’ve now seen that lossless sample compression, the ability to compress finite samples without loss of label inference, gives a simple algorithm for labeling an $n$-point dataset $X$ in $O(\log(n))$ queries while never storing more than $O(1)$ examples at a time. Furthermore, we’ve shown that lossless compression isn’t a hopelessly strong condition—basic real-world questions such as the odd-one-out query often lead to small compression schemes. In our recent paper we give a few more examples of this phenomenon for richer classes such as decision trees and halfspaces in 2D.
On the other hand, there is still much left to explore! Lossless sample compression gives a sufficient condition for bounded memory active learning, but it is not clear if the condition is necessary. The parameter is closely related to a necessary condition for active learning called inference dimension (see our previous post or KLMZ’s original paper for a description), and it is an open problem whether these two measures are equivalent. A positive resolution would imply that every actively learnable class is also actively learnable in bounded memory! Finally, it is worth noting that the techniques we discuss in this post are not robust to noise. Building a general framework for the more realistic noise-tolerant regime remains an interesting open question as well.
Footnotes
Note that this does not immediately imply a compression set for general samples. However, the definition of lossless compression can be weakened to allow for seperate compression schemes for positive and negative examples without affecting the resulting implications on bounded memory learnability. ↩
]]><a href='http://cseweb.ucsd.edu/~nmhopkin/'>Max Hopkins</a>Understanding Instance-based Interpretability of Variational Auto-Encoders2021-10-15T17:00:00+00:002021-10-15T17:00:00+00:00https://ucsdml.github.io//jekyll/update/2021/10/15/interpretability-vaeBackground: Instance-based Interpretability
Modern machine learning algorithms can achieve very high accuracy on many tasks such as image classification. Despite their great success, these algorithms are often black boxes as their predictions are mysterious to humans. For example, when we feed an image to a dog-versus-cat classifier, it says: “After a matrix product and max pooling and a non-linearity and a skip connection and another 100 math operations, look, the probability that ‘this image is a cat’ is 99%!” Unfortunately, this makes no sense to a human at all. To understand what is going on, we need information that can be easily interpreted by human. One way to provide more interpretable answer is to ask:
Which training samples are most responsible for the prediction of a test sample?
This is called a counterfactual question. Instance-based interpretation methods answer this question by designing an interpretability score between every training sample and the test sample. High scores imply importance. Then, we can interpret the prediction by saying: the classifier labels the test image as a cat because these other training samples are cats, and they are most responsible for the prediction of the test image.
The notion of influence functions is a popular instance-based interpretability method for supervised learning. The intuition is: if removing some $x$ in the training set results in a large difference of the prediction (such as the logits) of $z$, then $x$ is very important for the prediction of $z$. Imagine $z$ is a very special cat that is visually different from all training images except for one sample $x$. Then, $x$ has large influence over $z$ because removing $x$ probably leads to an incorrect prediction of $z$.
Interpretations for Unsupervised Learning
For supervised learning, instance-based interpretability methods reveal why a classifier makes a certain prediction. What about unsupervised learning? Our recent paper investigates this problem for several unsupervised learning methods. The first challenge is, how do we frame the counterfactual question in unsupervised learning?
When the model fits a probability density to the training data, we ask: which training samples are most responsible for increasing the log-likelihood of a test sample? In deep generative models such as variational auto-encoders (VAE), likelihood is not available. VAEs are optimized to maximize the evidence lower bound (ELBO) of the log-likelihood. Then, we ask: which training samples are most responsible for increasing the ELBO of a test sample?
Then, these questions can readily be answered by influence functions with proper loss functions. Formally, let $X=\{x_1,\cdots,x_n\}$ be the training set, and $\mathcal{A}$ be the unsupervised model. That is, $\mathcal{A}(X)$ returns the model fit to $X$. Let $L(X;\mathcal{A}) = \frac1N \sum_{i=1}^N \ell(x_i;\mathcal{A}(X))$ be the loss function, where the loss $\ell$ is negative log-likelihood in density estimators and negative ELBO in VAE. Then, the influence function of a training sample $x_i$ over a test sample $z$ is the difference of the losses at $z$ between models trained with and without $x_i$. Formally, we define the influence function as
\[\mathrm{IF}_{X,\mathcal{A}}(x_i,z) = \ell(z;\mathcal{A}(X\setminus\{x_i\})) - \ell(z;\mathcal{A}(X)).\]
We provide intuition for influence functions in the next section.
What Should Influence Functions Tell Us?
What does it mean if $\mathrm{IF}(x_i,z)\gg0$? Straightforward, we have $\ell(z;\mathcal{A}(X\setminus\{x_i\})) \gg \ell(z;\mathcal{A}(X))$, which means removing $x_i$ should result in a large increase of the loss at $z$. In other words, $x_i$ is very important for the model $\mathcal{A}$ to learn $z$. Similarly, if $\mathrm{IF}(x_i,z)\ll0$, then $x_i$ negatively impacts the model in learning $z$; and if $\mathrm{IF}(x_i,z)\approx0$, then $x_i$ hardly impacts it.
For conciseness, we call training samples that have positive / negative influences over a test sample $z$ proponents / opponents of $z$. In supervised learning, strong proponents and opponents of $z$ are very important to explain the model’s prediction of $z$. Strong proponents help the model correctly predict the label of $z$ because they reduce the loss at $z$, while strong opponents harm it because they increase the loss at $z$. Empirically, strong proponents of $z$ are visually its similar samples from the same class, while strong opponents of $z$ are usually its dissimilar samples from the same class or its similar samples from a different class.
In unsupervised learning, we expect that strong proponents increase the likelihood of $z$ and strong opponents reduce it, so we ask:
Which training samples are strong proponents and opponents of a test sample, respectively?
In particular, when we let $z = x_i$, we obtain a concept called self influence, or $\mathrm{IF}(x_i,x_i)$. This concept is very interesting in supervised learning because self influences provide rich information about memorization of training samples. For example, Feldman and Zhang study neural network memorization through the lens of self influences in this paper. Intuitively, high self influence samples are atypical, ambiguous or mislabeled, while low self influence samples are typical. We want to know what self influences reveal in unsupervised learning, so we ask:
Which training samples have the highest and lowest self influences, respectively?
By looking at these counterfactual questions, we hope to reveal what influence functions can tell us about (1) inductive biases of unsupervised learning models and (2) unrevealed properties of the training set (or distribution) such as outliers.
Intuitions from Classical Unsupervised Learning
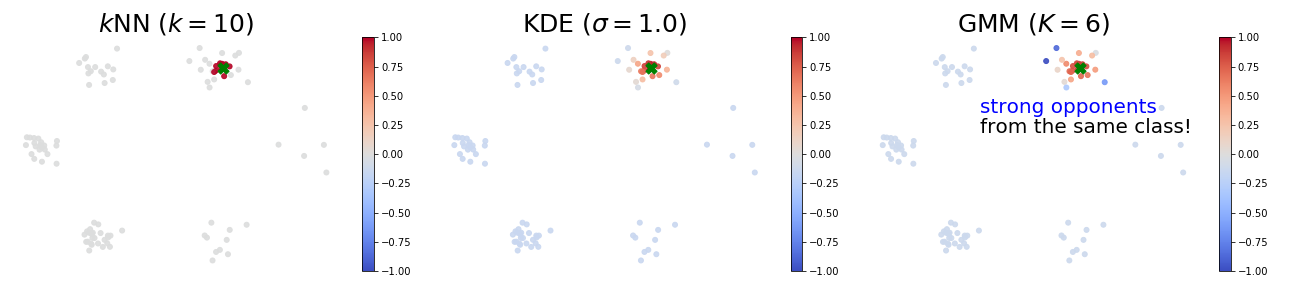
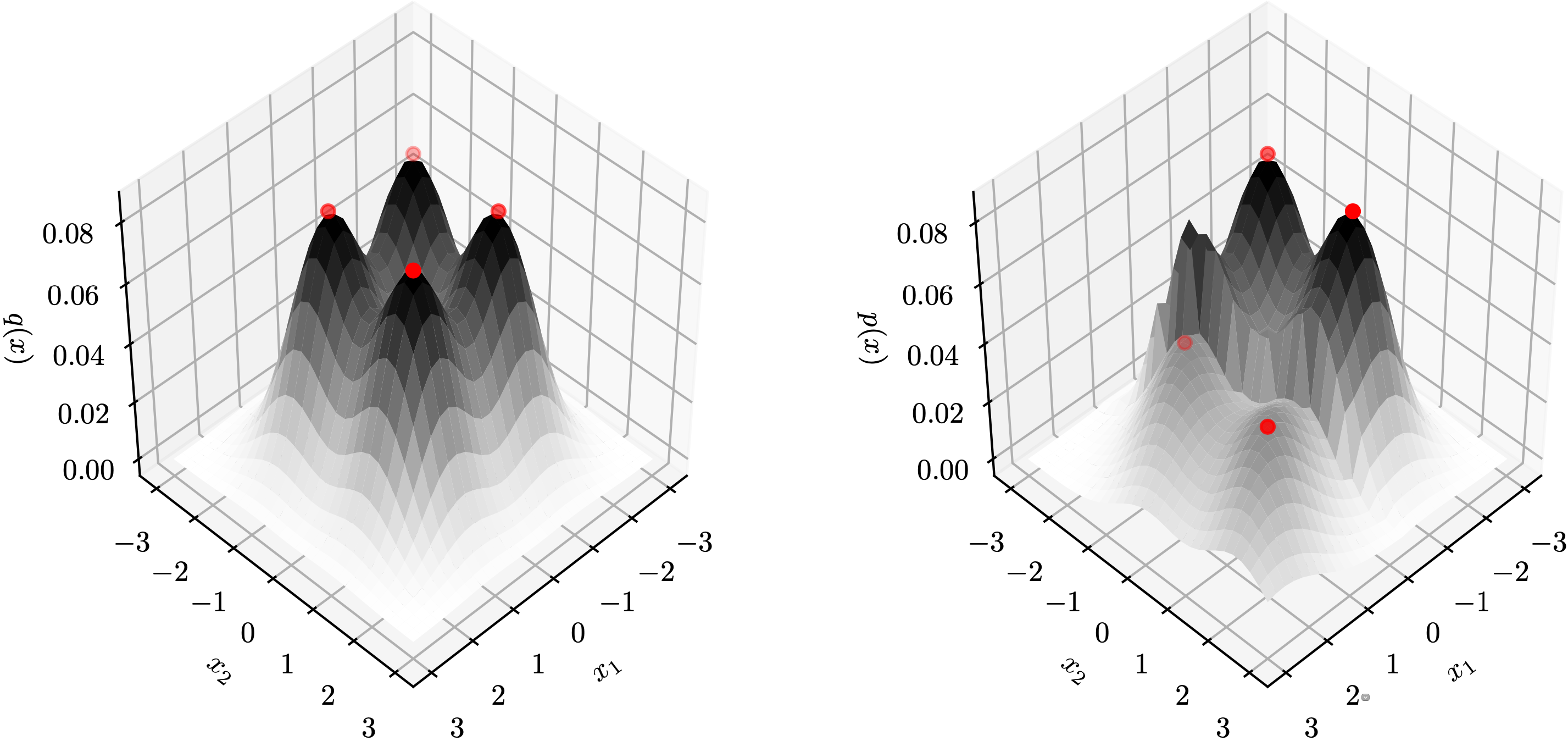
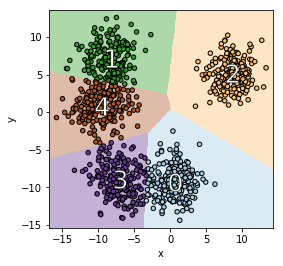
Let’s first look at these questions in the context of several classical unsupervised learning methods. The goal is to provide intuition on what influence functions should tell us in the unsupervised setting. Consider the following two-dimensional training data $X$ composed of six clusters.
The figure below provides some insights of high and low self influence samples. The color of a point represents its self influence (red means high and blue means low).
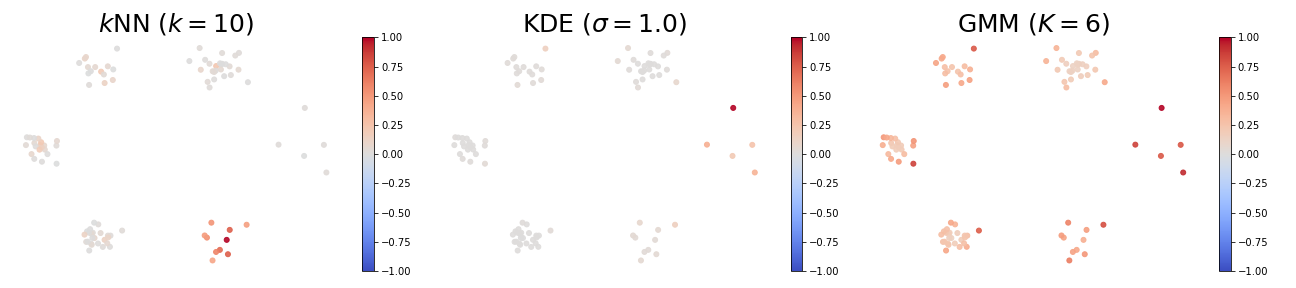
When using the $k$-NN density estimator, high self influence samples come from a cluster with exactly $k$ points.
When using the KDE density estimator, high self influence samples come from sparse regions, and low self influence samples come from dense regions.
When using the GMM density estimator, high self influence samples are far away to cluster centers, and low self influence samples are near cluster centers.
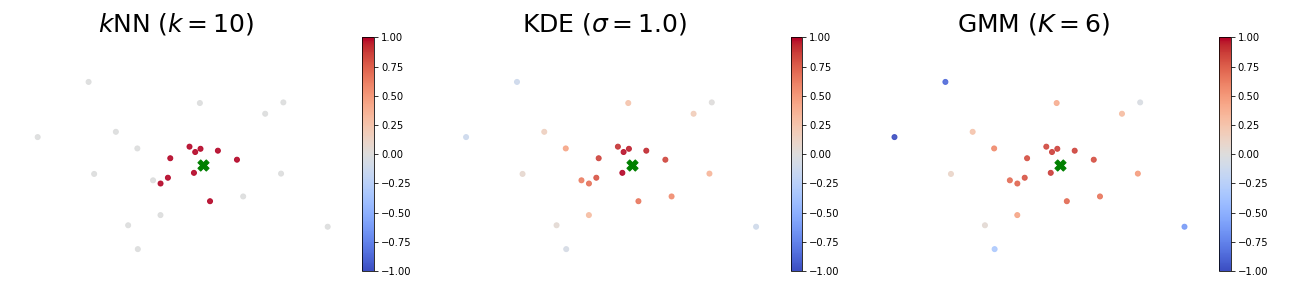
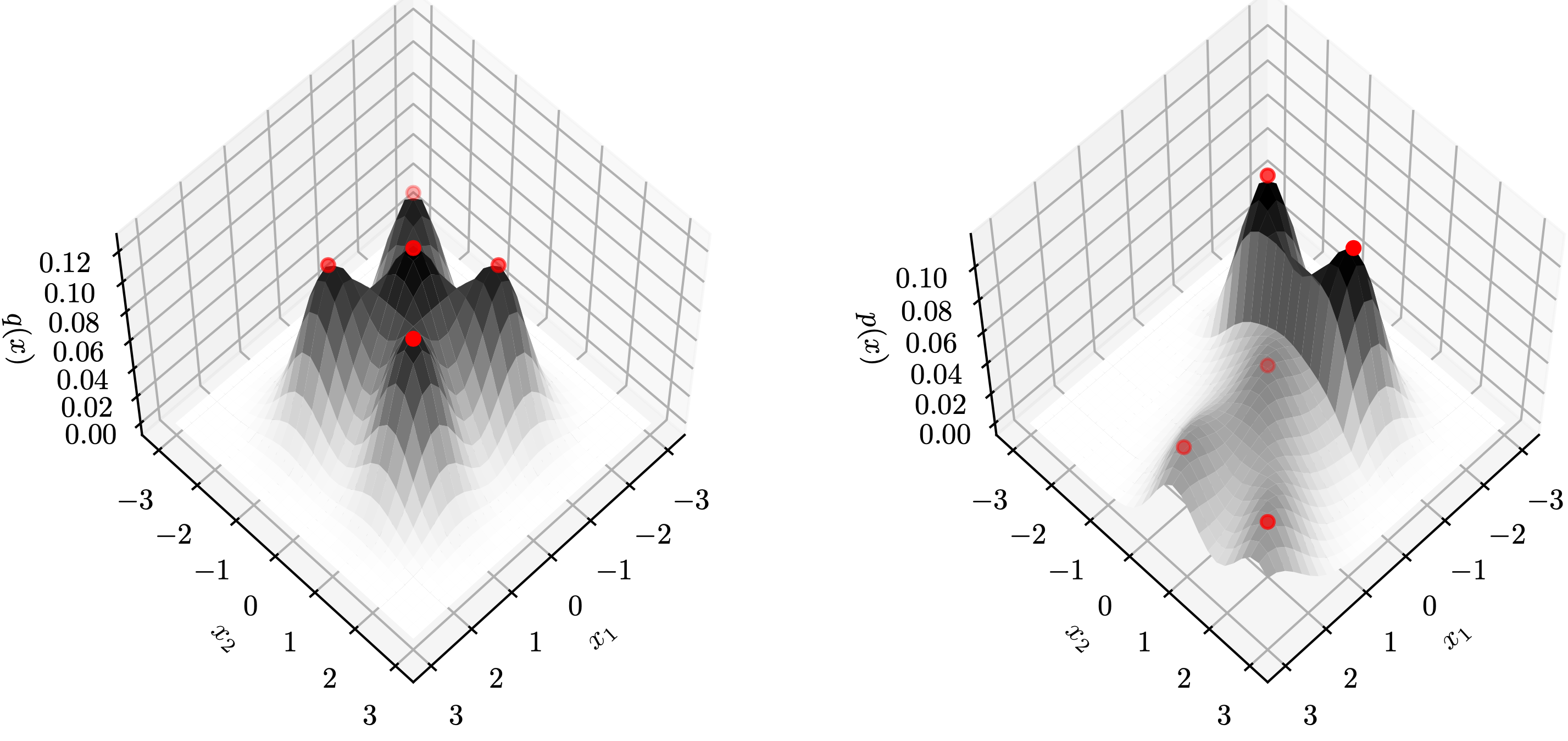
Proponents and Opponents
The figures below visualize an example of proponents and opponents. The test sample $z$ is marked as the green ✖︎ symbol, and the color of a point represents its influence over the test sample (red means proponents and blue means opponents). In all these models, strong proponents are the nearest neighbours of the test sample.
When using the $k$-NN or the KDE density estimator, strong proponents of $z$ are exactly its $k$ nearest neighbours.
KDE seems to be the soft version of $k$-NN: influences over $z$ gradually decrease as distances to $z$ increase.
When using the GMM density estimator, it is surprising to observe that some strong opponents (blue points) of $z$ are from the same cluster! This phenomenon indicates that removing a sample from the same class can possibly increase the likelihood at $z$. To see why this happens, we note the GMM is parametric and has limited capacity. Therefore, training samples that are far away to the cluster centers can largely affect the mean and covariance matrices of the learned Gaussians.
Scatter plots of influences of all training samples:
And the zoom in view that only shows the cluster which $z$ belongs to:
Note: please refer to Section 3 of our paper for the closed-form influence functions.
Variational auto-encoders are a class of generative models composed of two networks: the encoder, which maps samples to latent vectors, and the decoder, which maps latent vectors to samples. These models are trained to maximize the evidence lower bound (ELBO), a lower bound of log-likelihood.
There are two challenges when we investigate influence functions in VAE.
The influence function involves computing the loss at a test sample. However, the ELBO objective in VAE has an expectation term over the encoder, so it cannot be precisely computed.
Solution: we compute the empirical average of the influence function. We provide a probabilistic error bound on this estimate: if the empirical average is over $\mathcal{\Theta}\left(\frac{1}{\epsilon^2\delta}\right)$ samples, then with probability at least $1-\delta$, the error between the empirical and true influence functions is no more than $\epsilon$.
The influence function is hard to compute, as it requires inverting a large Hessian matrix. The number of rows in this matrix equals to the number of parameters in the VAE model, which can be as large as a million. Consequently, inverting this matrix (or even computing Hessian vector products) can be computationally infeasible.
Solution: we propose a computationally efficient algorithm called VAE-TracIn. It is based on the fast TracIn algorithm, an approximation to influence functions. TracIn is efficient because (1) it only involves computing the first-order derivative of the loss, and (2) it can be accelerated with only a few checkpoints.
A sanity check
Does VAE-TracIn find the most influential training samples? In a good instance-based
interpretation, training samples should have large influences over themselves. Therefore, we design the following sanity check (which is analogous to the identical subclass test by Hanawa et al. in this reference):
Are training samples the strongest proponents over themselves?
The short answer is: yes. We visualize some training samples and their strongest proponents in the figures below. A sample is marked in a green box if it is exactly its strongest proponent, and in a red box otherwise. Quantitatively, almost all ($>99\%$) training samples are the strongest proponents of themselves, with only very few exceptions. And as shown, even if a samples is not its strongest proponent, it still ranks very high in the order of influence scores.
Self influences for VAEs
We visualize high self influence samples below. We find these samples are either hard to recognize or visually high-contrast.
We then visualize low self influence samples below. We find these samples share similar shapes or background.
These findings are consistent with the memorization analysis in the supervised setting by Feldman and Zhang in this reference. Intuitively, high self influence samples are very different from most samples, so they must be memorized by the model. Low self influence samples, on the other hand, are very similar to each other, so the model does not need to memorize all of them. Quantitatively, we also find self influences correlate to the loss of training samples: generally, the larger loss, the larger self influence.
The intuition on self influences leads to an application in unsupervised data cleaning. Because high self influence samples are visually complicated and different, they are likely to be outside the data manifold. Therefore, we can use self influences to detect unlikely (noisy, contaminated, or even incorrectly collected) samples. For example, they could be
unrecognizable handwritten digits or objects in MNIST or CIFAR. Similar approaches in supervised learning use self influences to detect mislabeled data or memorized samples.
Proponents and Opponents in VAEs
We visualize strong proponents and opponents of several test samples below.
In MNIST, many strong proponents and opponents of test samples are similar samples from the same class. Especially, strong proponents look very similar to test samples, and strong opponents are visually slightly different. For example, the opponents of the test “two” have very different thickness and styles. Quantitatively, $\sim 80\%$ of the strongest proponents and $\sim 40\%$ of the strongest opponents have the same label as test samples. In addition, both of them have small latent space distance to the test samples. One can find this is very similar to GMM.
In CIFAR, we find strong proponents seem to match the color of the images – including the background and the object – and they tend to have the same but brighter colors. Strong opponents, on the other hand, tend to have very different colors as the test samples. Quantitatively, strong proponents have large norms in the latent space, indicating they are very likely to be outliers, high-contrast samples, or very bright samples. This observation is also validated in the visualizations. One can further connect this observation to influence functions in supervised learning. Hanawa et al. find extremely large norm samples are selected as relevant instances by influence functions in this reference, and Barshan et al. find large norm samples can impact a large region in the data space when using the logistic regression in this reference.
Open Questions
There are many open questions based on our paper. Here is a list of some important future directions.
How to design efficient instance-based interpretation methods for modern, large unsupervised learning models trained on millions of samples?
How can we use the instance-based interpretations to detect biases and fairness in models and data?
What are the other applications of instance-based interpretation methods?
]]><a href='https://cseweb.ucsd.edu/~z4kong'>Zhifeng Kong</a> and <a href='http://cseweb.ucsd.edu/~kamalika'>Kamalika Chaudhuri</a>Connecting Interpretability and Robustness in Decision Trees through Separation2021-09-24T17:00:00+00:002021-09-24T17:00:00+00:00https://ucsdml.github.io//jekyll/update/2021/09/24/interpretable-robust-treesTL;DR We construct a tree-based model that is guaranteed
to be adversarially robust, interpretable, and accurate.
Imagine a world where computers are fully integrated into
our everyday lives. Making decisions independently, without
human intervention. No need to worry about overly exhausted
doctors making life-changing decisions or driving your car
after a long day at the office. Sounds great, right? Well,
what if those computers weren’t reliable? What if a
computer decided you need to go through surgery without
telling you why? What if a car confused a child with a
green light? It doesn’t sound so great after all.
Before we fully embrace machine learning, it needs to be reliable.
The cornerstones for reliable machine learning are (i) interpretability,
where the model’s decisions are transparent, and (ii) robustness, where small changes
to the input do not change the model’s prediction.
Unfortunately, these properties are generally studied in isolation or only empirically.
Here, we explore interpretability and robustness simultaneously,
and examine it both theoretically and empirically.
We start this post by explaining what we mean by interpretability and robustness.
Next, to derive guarantees, we need some assumptions on the data.
We start with the known $r$-separated data.
We show that although there exists a tree that is accurate and robust,
such tree can be exponentially large, which makes it not interpretable.
To improve the guarantees, we make a stronger assumption on the data
and focus on linearly separable data.
We design an algorithm called BBM-RS and prove that it is accurate, robust, and interpretable on
linearly separable data.
Lastly, real datasets may not be linearly separable, so to understand how BBM-RS performs in practice,
we conduct an empirical study on $13$ datasets.
We find out that BBM-RS brings better robustness and interpretability while performing competitively
on test accuracy.
What do we mean by interpretability and robustness?
Interpretability
A model is interpretable if the model is simple and self-explanatory.
There are several forms of
self-explanatory models,
e.g., decision sets,
logistic regression, and
decision rules.
One of the most fundamental interpretable models, which we focus on here, are
small decision trees.
We use the size of a tree to determine whether it is interpretable or not.
Robustness
We also want our model to be robust to adversarial perturbations.
This means that if example $x$ is changed, by a bit, to $x’$, the model’s
answer remains the same.
By “a bit”, we mean that $x’=x+\delta$ where $\|\delta\|_\infty\leq r$ is
small. A model $h:\mathbf{X} \rightarrow \{-1, +1\}$ is robust at $x$ with radius
$r$ if for all such $x’$ we have that $h(x)=h(x’)$. The notion of astutenesswas previously introduced
to jointly measure the robustness and the accuracy of a model.
The astuteness of a model $h$ at radius $r > 0$ under a distribution $\mu$
is \[\Pr_{(x,y)\sim\mu}[h(x’)=y \ |\ \forall x’ \text{ with } \|x-x’\|_\infty\leq r].\]
Guarantees under different data assumptions
Without any assumptions on the data, we cannot guarantee
accuracy, interpretability, and robustness to hold simultaneously.
For example, if the true labels of the examples are different for close
examples, a model cannot be astute (accurate and robust).
In this section, we explore which data properties are sufficient for astuteness and interpretability.
$r$-Separation
A prior work suggested
focusing on datasets that satisfy $r$-separation.
A binary labeled data distribution is $r$-separated if every two differently labeled examples, $(x^1,+1)$,$(x^2,-1)$, are far apart,
$\|x^1-x^2\|_\infty\geq 2r.$
Yang et al. showed that
$r$-separation is sufficient for robust learning.
Therefore, we examine whether it is also sufficient for accuracy and
interpretability.
We have two main findings.
First, we found that there is a accurate decision tree with size
independent of the number of examples.
Second, we discovered that the size of the accurate tree can be exponential
in the number of features.
Combining these two findings, it appears we need to find a stronger assumption on the data to
be able to have guarantees on both accuracy and interpretability.
Linear separation
Next, we investigate a stronger assumption — linear separation with a
$\gamma$-margin.
Intuitively, it means that a hyperplane separates the two labels in the data,
and the margin (distance of the closest point to the hyperplane) is at
least $\gamma$ (larger $\gamma$ means larger margin for the classifier).
More formally, there exists a vector $w$ with $\|w\|_1=1$ such that
for each training example and its label $(x, y)$, we have
$ywx\geq \gamma$.
Linear separation is a popular assumption in the research of
machine learning models, e.g., for
support vector machines,
neural networks,
and decision trees.
Using a generalization of
previous work,
we know that under the linear separation assumption, there has to be a feature
that gives nontrivial information.
To formalize it, we use the notion of
decision stumps and
weak learners.
A decision stump is a (simple) hypothesis of the form $sign(x_i-\theta)$ defined
by a feature $i$ and a threshold $\theta$.
A hypothesis class is a $\gamma$-weak learner if one can learn it with accuracy
$\gamma$ (slightly) better than random, i.e., if there is always a
hypothesis in the class with accuracy of at least $1/2+\gamma$.
Now, we look at the hypothesis class of all possible decision stumps, and we want
to show that this class is a weak learner.
For each dataset $S=((x^1,y^1),\ldots,(x^m,y^m))$, we denote
the best decision stump for this dataset by $h_S(x)=sign(x_i-\theta)$, where $i$
is a feature and $\theta$ is a threshold that minimize the error
$\sum_{j=1}^m sign(x^j_i < \theta) y^j.$
We can show that $h_S$ has accuracy better than $0.5$, i.e., better than a
random guess:
Fix $\alpha>0$.
For any distribution $\mu$ over $[-1,+1]^d\times\{-1,+1\}$ that satisfies
linear separability with a $\gamma$-margin, and for any $\delta\in(0,1)$ there
is $m=O\left(\frac{d+\log\frac1\delta}{\gamma^2}\right)$, such that with
probability at least $1-\delta$ over the sample $S$ of size $m$, it holds that
$$\Pr_{(x,y)\sim\mu}(h_S(x)=y)\geq \frac12+\frac{\gamma}{4}-\alpha.$$
This result proves that there exists a classifier $h_S$ in the hypothesis class of
all possible decision stumps that produces a non-trivial
solution under the linear separability assumption.
Using this theorem along with the result from
Kearns and Mansour,
we can show that
CART-type
algorithms can deliver a small tree with high accuracy.
As a side benefit, this is the first time that a distributional
assumption that does not include feature independence is used.
Many papers on theoretical guarantees of decision trees assumed either uniformity or feature independence
(papers 1,
2, and
3).
Are we done? Is this model also robust?
New algorithm: BBM-RS
Designing robust decision trees is inherently a difficult task.
One reason is that, generally, the models defined by the right and left subtrees
can be completely different.
The feature $i$ in the root determines if the model
uses the right or left subtree.
Thus, a small change in the $i$-th feature completely changes the model.
To overcome this difficulty, we focus on a specific class of decision trees.
Risk score
We design our algorithm to learn a specific kind of decision tree —
risk score.
A risk score is composed of several conditions (e.g., $age \geq 75$), and each
is matched with an integer weight.
A score $s(x)$ of example $x$ is the weighted sum of all the satisfied
conditions.
The label is then $sign(s(x))$.
An example of the risk score model on the bank dataset.
Each satisfied condition is multiplied by its weight and summed. Bias term is always satisfied.
If the total score $>0$, the risk model predicts "1" (i.e., the client will open a bank account after a marketing call).
All features are binary (either $0$ or $1$).
For a concrete example, a person with age greater than 75, called before but the previous call
was not successful, and the consumer price index is greater than 93.5, the total score would be
$1$ and the prediction would be "1".
features
weights
Bias term
-5
+ ...
Age $\geq 75$
2
+ ...
Called before
4
+ ...
Previous call was successful
2
+ ...
total scores=
A risk score can be viewed as a decision tree with the same feature-threshold pair at
each level (see example below).
A risk score has simpler structure than a standard decision tree,
and it generally has fewer number of unique nodes.
Hence, they are considered
more interpretable than decision trees.
The following table shows an example of a risk score.
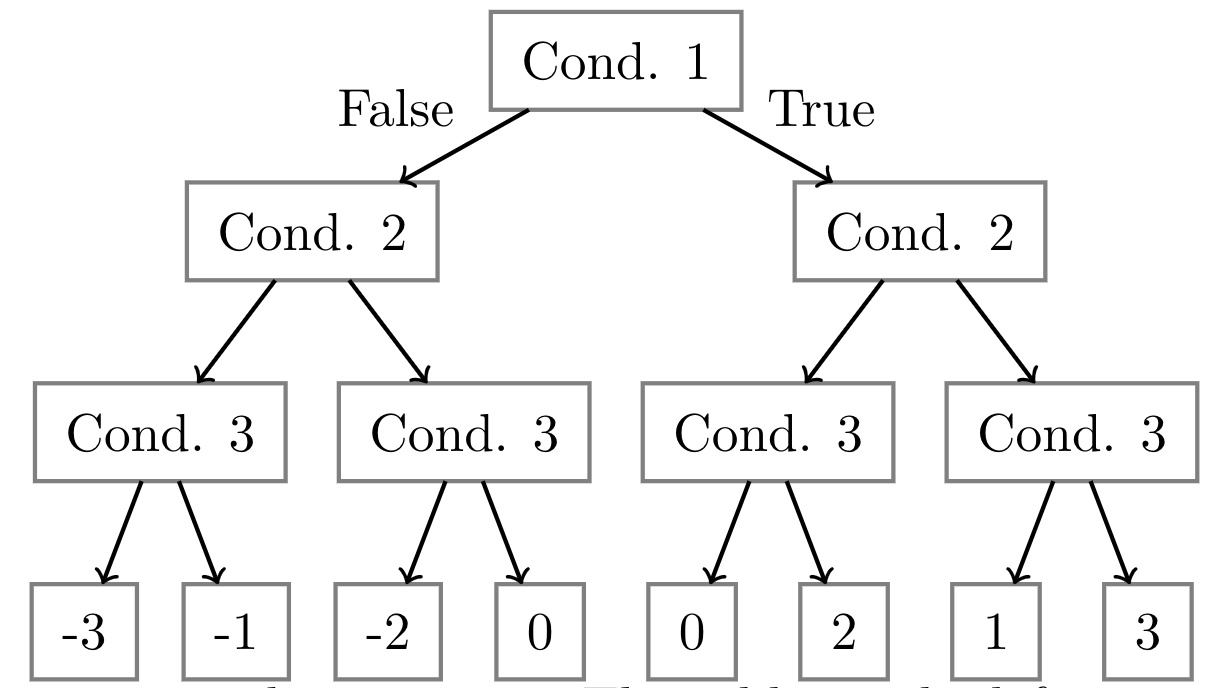
Here is an example of how to convert a risk score into a decision tree.
The table on the left is an example of a risk score that may be used by a doctor to determine
whether a patient caught a cold or not.
It has three conditions and the figure on the right is the corresponding decision tree.
For each node in the tree, the branch towards the right represents the path to take if the condition is true.
The leaves represent the final risk score of the given condition.
For a concrete example, if a patient has a fever, coughs, but does not sneeze, we would follow the green
path in the decision tree and result in a score of $2$.
features
weights
Bias term
-3
+ ...
Fever
3
+ ...
Sneeze
1
+ ...
Cough
2
+ ...
total scores=
BBM-RS
We design a new algorithm for learning risk scores by utilizing the known
boosting method
boost-by-majority
(BBM).
The different conditions are added to the risk score one by one, using
the weak learner.
BBM has the benefit of ensuring the weights in the risk score
are small integers.
This will lead to an interpretable model with size only
$O(\gamma^{-2}\log1/\epsilon)$ where the model has accuracy $1-\epsilon$.
Now we want to make sure that the risk model is also robust.
The idea is to add
noise.
We take each point in the sample and just make sure that it’s a little
bit closer to the decision boundary, see the figure below.
The idea is that if the model is correct for the noisy point, then it
should be correct for the point without the noise.
To formally prove it, we show that choosing the risk-score conditions in a specific
way ensures that they are monotone models.
In such models, adding noise in the way we described is
sufficient for robustness.
Before we examine this algorithm on real datasets, let’s check its running time.
We focus on the case the margin and desired accuracy are constants.
In this case, the number of steps BBM-RS will take is also constant.
In each step, we run the weak learner and find the best $(i,\theta)$.
So the overall time is linear (up to logarithmic factors) in the input size and the time to run the
weak learner.
To summarize, we designed a new efficient algorithm, BBM-RS, that is robust, interpretable, and
has high accuracy. The following theorem shows this. Please refer to
our paper for the pseudocode of BBM-RS
and more details for the theorem.
Suppose data is $\gamma$-linearly separable and fix $\epsilon,\delta\in(0,1)$.
Then, with probability $1-\delta$ the output of BBM-RS, after receiving
$(d+\log(1/\delta))\log(1/\epsilon)\gamma^{-O(1)}$ samples, has astuteness
$1-\epsilon$ at radius $\gamma/2$ and has $O(\gamma^{-2}\log(1/\epsilon))$
feature-threshold pairs.
Performance on real data
For BBM-RS, our theorem is restricted to linearly separable data.
However, real datasets may not perfectly linearly separable.
A straightforward question: is linear separability a reasonable
assumption in practice?
To answer this question, we consider $13$ real datasets (here we present the
results for four datasets; for more datasets, please refer to our
paper).
We measure how linearly separable each of these datasets is.
We define the linear separateness as one minus the minimal fraction
of points that needed to be removed for the data to be linearly separable.
Since finding the optimal linear separateness on arbitrary data
is NP-hard,
we approximate linear separateness with the training accuracy of the best linear classifier we can
find (since removing the incorrect examples for a linear classifier would make the dataset linearly
separable).
We train linear SVMs with different regularization parameters and record the best training accuracy.
After removing the misclassified points by an SVM, we are left with accuracy
fraction of linearly separable examples.
The higher this accuracy is, the more linearly separable the data is.
The following table shows the results and it reveals that most datasets
are very or moderately close to being linearly separated.
This indicates that the linear assumption in our theorem may not be too
restrictive in practice.
linear separateness
adult
0.84
breastcancer
0.97
diabetes
0.77
heart
0.89
Even though these datasets are not perfectly linearly separable, BBM-RS can
still be applied (but the theorem may not hold).
We are interested to see how BBM-RS performed against others on these
non-linearly separable datasets.
We compare BBM-RS to three baselines,
LCPA,
decision tree (DT), and
robust decision tree (RobDT).
We measure a model’s robustness by evaluating its
Empirical robustness (ER), which is the
average $\ell_\infty$
distance to the closest adversarial example on correctly predicted test examples.
The larger ER is, the more robust the classifier is.
We measure a model’s interpretability by evaluating its
interpretation complexity (IC).
We measure IC with the number of unique feature-threshold pairs in the
model (this corresponds to the number of conditions in the risk score).
The smaller IC is, the more interpretable the classifier is.
The following tables show the experimental results.
test accuracy (higher=better)
DT
RobDT
LCPA
BBM-RS
adult
0.83
0.83
0.82
0.81
breastcancer
0.94
0.94
0.96
0.96
diabetes
0.74
0.73
0.76
0.65
heart
0.76
0.79
0.82
0.82
ER (higher=better)
DT
RobDT
LCPA
BBM-RS
adult
0.50
0.50
0.12
0.50
breastcancer
0.23
0.29
0.28
0.27
diabetes
0.08
0.08
0.09
0.15
heart
0.23
0.31
0.14
0.32
IC feature threshold pairs (lower=better)
DT
RobDT
LCPA
BBM-RS
adult
414.20
287.90
14.90
6.00
breastcancer
15.20
7.40
6.00
11.00
diabetes
31.20
27.90
6.00
2.10
heart
20.30
13.60
11.90
9.50
From the tables, we see that BBM-RS has a test accuracy comparable to other
methods.
In terms of robustness, it performs slightly better than others (performing the
best on three datasets among a total of four).
In terms of interpretability, BBM-RS
performs the best in three out of four datasets.
All in all, we see that BBM-RS can bring better robustness and interpretability
while performing competitively on test accuracy.
This shows that BBM-RS not only performs well theoretically, it also performs
well empirically.
Conclusion
We investigated three important properties of a classifier: accuracy, robustness, and
interpretability.
We designed and analyzed a tree-based algorithm that provably achieves all these properties, under
linear separation with a margin assumption.
Our research is a step towards building trustworthy models that provably achieve many desired
properties.
Our research raises many open problems.
What is the optimal dependence between accuracy, interpretation complexity,
empirical robustness, and sample complexity?
Can we have guarantees using different notions of interpretability?
We showed how to construct an interpretable, robust, and accurate model. But,
for reliable machine learning models, many more properties are required,
such as privacy and fairness.
Can we build a model with guarantees on all these properties simultaneously?
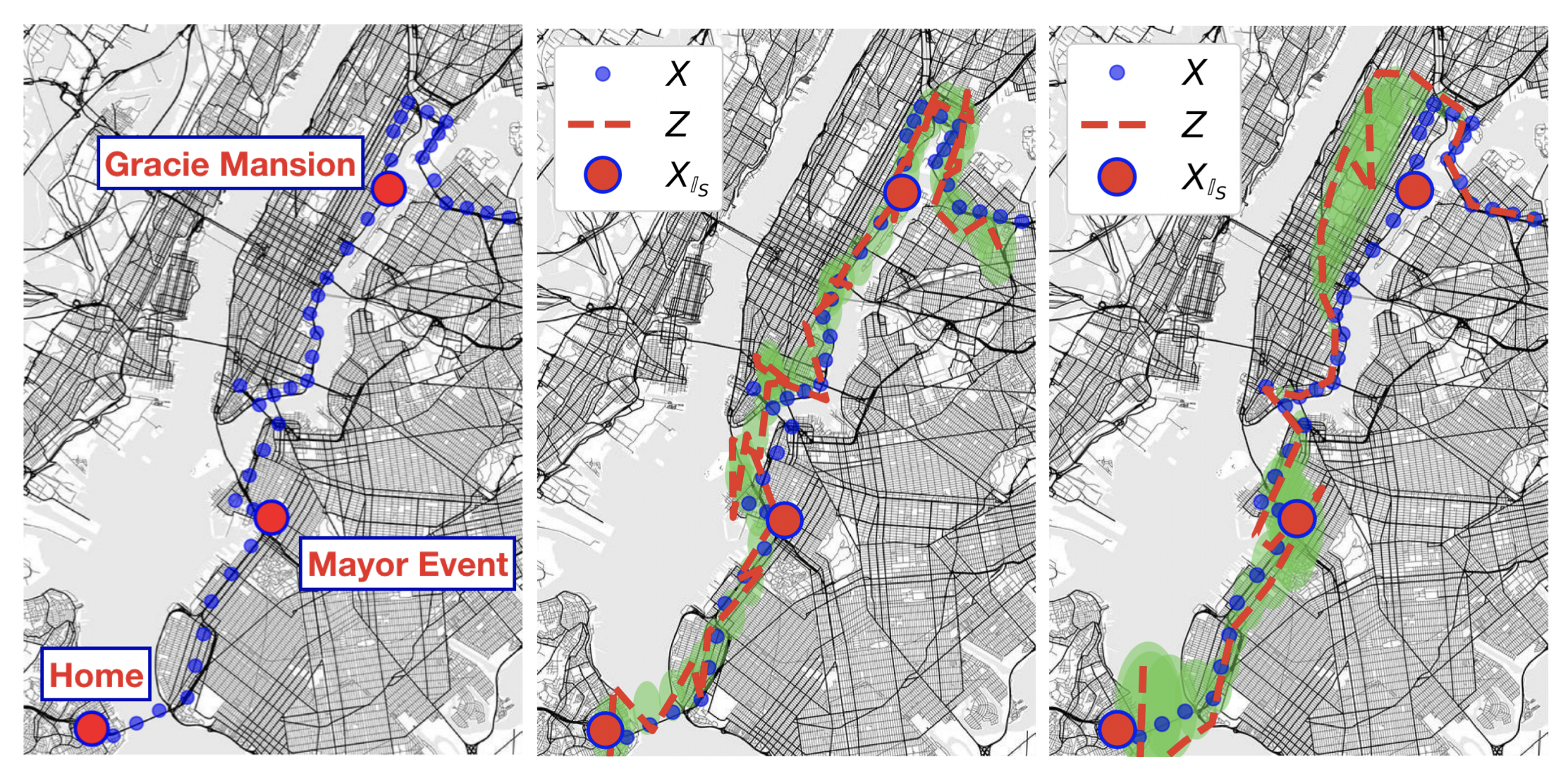
]]><a href='https://sites.google.com/view/michal-moshkovitz'>Michal Moshkovitz</a> and <a href='http://yyyang.me'>Yao-Yuan Yang</a>Location Trace Privacy Under Conditional Priors2021-05-10T19:00:00+00:002021-05-10T19:00:00+00:00https://ucsdml.github.io//jekyll/update/2021/05/10/location-trace-privacyImagine a mobile app that repeatedly records your geolocation over a short period of time – say a day. We call this sequence of locations a location trace. Ideally, the app would like to use these locations to send recommendations or ads or even reminders. But there is the issue of privacy; many people including myself, would feel uncomfortable if our exact locations were to be shared with and recorded by apps. One option may be to completely shut off all location services. But is it possible to have a happy medium? In other words, can we obscure a location trace of an user while still providing some privacy?
Rigorous Privacy Definitions: Differential and Inferential
Before we get to what privacy means in this case, let us look at how rigorous privacy definitions work. Broadly speaking, the literature has two main philosophies of rigorous definitions of statistical privacy — differential and inferential privacy. Differential privacy is an elegant privacy definition designed by cryptographers Cynthia Dwork, Frank McSherry, Kobbi Nissim and Adam Smith in 2006. The philosophy here is that the participation of a single person in the data should not make a big difference to the probability of any outcome; this, in turn, implies that an adversary watching the output of a differentially private algorithm cannot determine for sure if a certain person is in the dataset or not. Differential privacy has many elegant properties — such as, robustness to auxiliary information, graceful composition and post processing invariance.
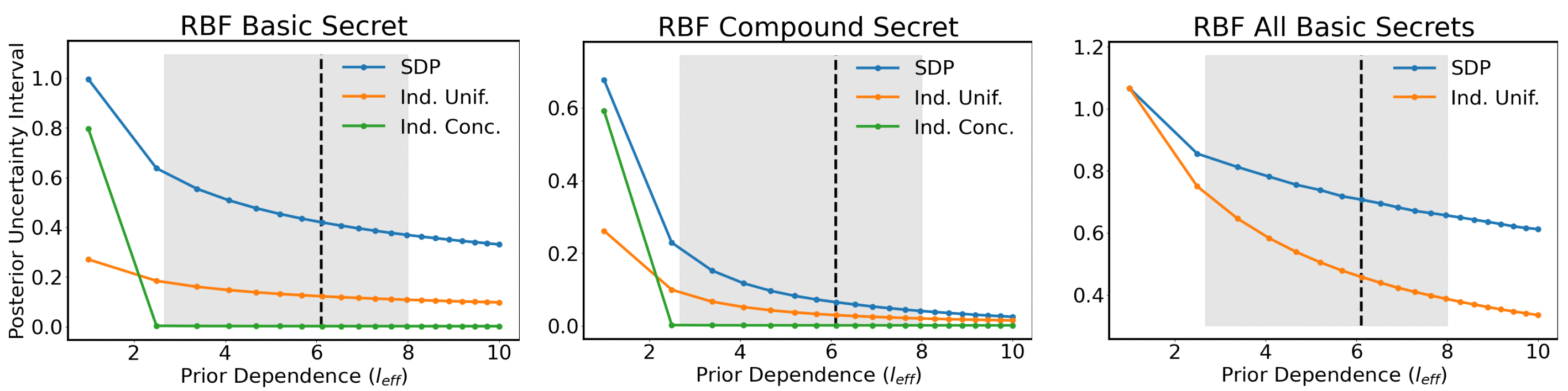
Inferential privacy in contrast means that an adversary with a certain prior knowledge does not gain a lot of extra knowledge after seeing the output of a private algorithm. While this notion is older than differential privacy, it was formalized by Kifer and Machanavajjhala in 2012 as the Pufferfish privacy framework. Inferential privacy does not always have the elegant properties of differential privacy, but it tends to be more flexible in the sense that it can obscure some specific events. Besides, some inferential privacy frameworks or algorithms do have graceful composition and are robust to certain kinds of auxiliary information. There is a no-free-lunch theorem that states that inferential privacy against all manner of auxiliary information will imply no utility — and so there is a limit to how far this can extend.
A Privacy Framework for Location Traces
Coming back to the privacy of location traces, let us now think about some options on how to model them in a rigorous privacy framework. There are two interesting aspects about location traces. First, location is continuous spatial data — and for both privacy and utility, we may need to obscure it up to a certain distance. We call this the spatiality aspect. But the more challenging aspect is correlation. My location at 10am is highly correlated with my location at 10:05, and not building this into a privacy framework may lead to privacy leaks.
Our first option is to use local differential privacy (LDP), which is basically differential privacy applied to a single person’s data. This will mean that two traces — one in New York and one in California — will be almost indistinguishable. However, this involves adding considerable noise to each trace — so much so as to render them completely useless. We will have very good privacy, but almost no utility whatsoever.
Our second option is to realize that while most people may be uncomfortable sharing fine-grained location information, they may be okay with coarse-grained data. For example, since I work at UCSD, which is in La Jolla, CA, I may not mind someone knowing that I spend most of my working hours in La Jolla; but I would not want them to know my precise location. This is known as geo-indistinguishability, and is achieved by adding independent noise with a radius $r$ to each location. This improves utility, if we are releasing a single location, but still has challenges with traces. If we average the private locations at 10am and 10:05am, then we get a better estimate since the underlying true locations are highly correlated.
Tradeoffs of three privacy definitions for location data: While DP prevents use of correlation, it does not allow for utility with individual traces. Geoindistinguishability works well for a single location, but cannot prevent an adversary from correlating points close by in time. Our definition (conditional inferential privacy) provides an intermediate: prevent inference against a class of priors while still offering valuable utility.
Conditional Inferential Privacy
This brings us to our framework, Conditional Inferential Privacy (CIP). Here we aim to obscure each location to within a radius $r$, while taking into account correlation across time through a Gaussian Process prior. Gaussian processes effectively model a sequence of $n$ random variables as an $n$-dimensional vector drawn from a multivariate normal distribution (see Rasmussen Ch. 2 for more detail). In the location setting, the correlation between two locations increases with their proximity in time. Gaussian processes are frequently used to model trajectories (Chen ‘15, Liang & Hass ‘03, Liu ‘98, Kim ‘11), so this serves as a good model for a prior. Through directly modeling correlations, we can ensure that we can obscure locations up to a radius $r$, even in the presence of these correlations.
Formally, our framework builds upon the Pufferfish inferential privacy framework. We have a set of basic secrets $S$ consisting of events $s_{x,t}$, which denotes “User was at location $x$ at time $t$”. These are the kinds of events that we would like to hide. In practice, we may choose to hide more complicated events — such as “User was at home at 10am and at the coffee shop at 10:05am”; these are modeled by a set of compound events $C$, which is essentially a set of tuples of the form $(s_{x_1, t_1}, s_{x_2, t_2}, …)$.
We then have the set of secret pairs $P$ which is a subset of $C \times C$ — these are the pairs of secrets that the adversary should not be able to distinguish between. Finally we have a set of priors $\Theta$, which is a set of Gaussian processes that presumably represents the adversary’s prior.
A mechanism $M$ is said to follow $(P, \Theta)$-CIP with parameters $(\lambda, \epsilon)$, if for all $\theta \in \Theta$ and all tuples in $(s, s’) \in P$, we have that:
\[D_{\text{Renyi}, \lambda} \Big(\Pr(M(X) = Z | s, \theta ) , \Pr(M(X) = Z | s’, \theta)\Big) \leq \epsilon\]
where $D_{\text{Renyi}, \lambda}$ is the Renyi divergence of order $\lambda$ (see Mironov ‘17 for background on Renyi divergence and its use in the privacy literature). Essentially what this means is that the distributions of the output of the mechanism $M$ are similar under the secret s and s’. Similar here means low Renyi divergence.
There are a couple of interesting things to note here. First, note that unlike differential privacy, here the privacy is over both the prior and the randomness in the mechanism; this is quite standard for inferential privacy. Second, observe that we use Renyi divergence in the definitions instead of the probability ratios or max divergence that is used in the standard differential privacy and Pufferfish privacy definition. This is because Renyi divergences have a natural synergy with Gaussians and Gaussian processes, which we use as our priors and mechanisms.
While not as elegant as differential privacy, this definition also has some good properties. We can show that we can get graceful decay of privacy for two trajectories of the same person from different time intervals — which is analogous to what is called parallel composition in the privacy literature. We also show that there is some robustness to side information. Details are in our paper.
Example of how CIP maintains high uncertainty at secret locations (times). Left: a real location trace unknowningly collected from an NYC mayoral staff member by apps on their phone. The red dots indicate sensitive locations. Middle: demonstration of how Geoindistinguishability (adding independent isotropic gaussian noise to each location, as in the red trace) allows for high certainty of true location by correlation. The green envelope shows the posterior uncertainty of a Bayesian adversary with a Gaussian process prior (a GP adversary). Right: demonstration of how a CIP mechanism efficiently thwarts the same adversary's posterior at sensitive locations, given the same utility budget. The mechanism achieves this by both concentrating the noise budget near sensitive locations and by strategically correlating noise added.
Related Work
It is worth noting that we are in no way the first to attempt to offer meaningful location privacy. However, our method is distinguished in that it works in a continuous spatiotemporal domain, offers local privacy within a radius $r$ for sensitive locations, and has a semantically meaningful inferential guarantee. A mechanism offered by Bindschaedler & Shokri releases synthesized traces satisfying the notion of plausible deniability, but this is distinctly different from providing a radius of privacy in the local setting, as we do. Meanwhile, the frameworks proposed by Xiao & Xiong (2015) and Cao et al. (2019) nicely characterize the risk of inference in location traces, but use only first-order Markov models of correlation between points, do not offer a radius of indistinguishability as in this work, and are not suited to continuous-valued spatiotemporal traces.
Results
With the definition in place, we can now measure the privacy loss of different mechanisms. The most basic mechanism is to add zero-mean isotropic Gaussian noise with equal standard deviation to every location in the trace and publish the result; if the added noise has standard deviation $\sigma$, then we can calculate the privacy loss under CIP, as well as the mean square error utility. If a certain utility is desired, we can calibrate $\sigma$ to it and obtain a certain privacy loss.
A more sophisticated mechanism is to add zero-mean Gaussian noise with different covariances to locations at different time points. It turns out that we can choose the covariances to minimize privacy loss for a given utility, and this can be done by solving a Semi-Definite Program. The derivation and more details are in our paper.
We provide below a snap-shot of what our results look like. On the x-axis, we are plotting a measure of how correlated our prior is. If the prior is highly correlated, then it is easy to leak privacy for mechanisms that add noise — and hence correlated priors are worse for privacy. On the y-axis, we are plotting the posterior confidence interval size of the adversary — higher means higher privacy. Both mechanisms are calibrated to the same mean-square error, and hence the privacy-utility tradeoff is better if the y-axis is higher. From the figure, we see that our SDP-based mechanism does lead to a better privacy-utility tradeoff, and as expected, privacy offered declines as the correlations grow worse.
Our inferentially private mechanism (blue line) maintains higher posterior uncertainty for a Bayesian adversary with a Gaussian process prior (a GP adversary) as compared to two Geoindistinguishability-based baselines (orange and green). The x-axis indicates the degree of correlation anticipated by the GP adversary. The left panel shows the posterior uncertainty for a single basic secret. The middle panel shows uncertainty for a compound secret. The right panel shows posterior uncertainty when we design our mechanism to maintain privacy at every location (all basic secrets). The gray window shows a range of realistic degrees of dependence (correlation) gathered from human mobility data.
Examples of the noise covariance chosen by our mechanism: Each frame is a covariance matrix optimized by our SDP mechanism to thwart inference at either a single location basic secret or a compound secret of two locations. Noise drawn from a multivariate normal with this covariance is added along the 50 point trace. The two frames on the left show covariance chosen to thwart a GP prior with an RBF kernel. The two frames on the right show covariance chosen to thwart a GP prior with a periodic kernel.
Conclusion
In conclusion, we take a stab at a long-standing challenge in offering location privacy — temporal correlations — and we provide a way to model them cleanly and flexibly through Gaussian Process priors. This gives us a way to quantify the privacy loss for correlated location trajectories and devise new mechanisms for sanitizing them. Our experiments show that our mechanisms offer better privacy-accuracy tradeoffs than standard baselines.
There are many open problems, particularly in the space of mechanism design. Can we improve the privacy-utility tradeoff offered by our mechanisms through other means, such as subsampling the traces or interpolation? Can we make our definition and our methods more robust to side information? Finally, location traces are only one example of correlated and structured data; a remaining challenge is to build upon the methodology developed here to design privacy frameworks for more complex and structured data.
]]><a href='https://cseweb.ucsd.edu/~kamalika/'>Kamalika Chaudhuri</a> and Casey MeehanThe Expressive Power of Normalizing Flow Models2020-11-16T17:00:00+00:002020-11-16T17:00:00+00:00https://ucsdml.github.io//jekyll/update/2020/11/16/expressive-power-normalizing-flowsBackground: Generative Models and Normalizing Flows
Generative models are one kind of unsupervised learning model in machine learning. Given a set of training data – such as pictures of dogs, audio clips of human speakers, and articles from certain websites – a generative model aims to generate samples that look/sound like they are samples from the dataset, but are not exactly any one of them. We usually train a generative model by maximizing the probability, or likelihood, of the samples under the model.
To understand complicated training data, generative models usually use very large neural networks (so they are also called deep generative models). Popular deep generative models include generative adversarial networks (GANs) and variational autoencoders (VAEs), which have achieved the state-of-the-art performances on most generative tasks. Below are examples showing that styleGAN (left) and VQ-VAE (right) can generate amazing high resolution images!
One might ask: as we already have powerful generative models, is everything done? No! There are many aspects in which we want to improve these models. Below are two points related to this blog.
First, we want to compute exact likelihood if possible. Both GANs and VAEs generate samples by applying a neural network transformation on a latent random variable $z$, which is usually a Gaussian. In this case, the sample likelihood cannot be exactly computed because complicated neural networks may map different $z$’s to the same output.
This is the reason why normalizing flows (NFs) were proposed. An NF learns an invertible function $f$ (which is also a neural network) to convert a source distribution, such as a Gaussian, to the distribution of the training data. Since $f$ is invertible, we can precisely compute the likelihood through the change-of-variable formula! This post includes the detailed math of the computation. Different from the decoder in VAEs and the generator in GANs (which usually transform a lower dimensional latent variable to the data distribution), the NF $f$ keeps the data dimension and $f^{-1}$ can map a sample back to the source distribution.
Second, we want a theoretical guarantee that these deep generative models are potentially able to learn an arbitrarily complicated data distribution. Without such theory, an empirically successful generative model might fail in another scenario, and we don’t want this risk to always exist! Despite its importance, this problem is super challenging due to the complicated structure of neural networks. For example, this paper analyzes GANs in transforming between very simple distributions.
This blog addresses the above two points by making a theoretical analysis to NFs. We provide a theoretical guarantee for NFs on $\mathbb{R}$ and some negative (impossibility) results for NFs on $\mathbb{R}^d$ where the dimension $d>1$.
Structure of Normalizing Flows
In general, to model complex training data like images, the normalizing flow $f$ needs to be a very complicated function. In practice, $f$ is usually constructed via a sequence of simple, invertible transformations, which we call base flow layers. The figure below illustrates the middle stages within the transformation from a simple source distribution to a complicated target distribution (figure from this link).
Examples of base flow layers include
planar layers: $f_{\text{pf}}(z)=z+uh(w^{\top}z+b)$, where $u,w,z\in\mathbb{R}^d,b\in\mathbb{R}$;
radial layers: $f_{\text{rf}}(z)=z+\frac{\beta}{\alpha+\|z-z_0\|}(z-z_0)$, where $z,z_0\in\mathbb{R}^d,\alpha,\beta\in\mathbb{R}$;
Sylvester layers: $f_{\text{syl}}(z)=z+Ah(B^{\top}z+b)$, where $A,B\in\mathbb{R}^{d\times m}, z\in\mathbb{R}^d, b\in\mathbb{R}^m$;
and Householder layers: $f_{\text{hh}}(z)=z-2vv^{\top}z$, where $v,z\in\mathbb{R}^d, v^{\top}v=1$.
The number of layers is usually very large in practice. For instance, in the MNIST dataset experiments, this paper uses 80 planar layers, and this paper uses 16 Sylvester layers.
Defining the Expressivity of Normalizing Flows
The invertibility of NFs may hugely restrict their expressive power, but to what extent? Our recent paper analyzes this through the following two questions:
Q1 (Exact transformation): Under what conditions is it possible to exactly transform the source distribution $q$ (e.g., a standard Gaussian) into the target distribution $p$ with a finite number of base flow layers?
Q2 (Approximation): Since sometimes exact transformation may be hard, when is it possible to approximate the target distribution $p$ in total variation distance? Do we need an incredibly large number of layers?
Our findings:
If $p$ and $q$ are defined on $\mathbb{R}$, then universal approximation can be achieved. That is, we can always transform $q$ to be arbitrarily close to any $p$.
If $p$ and $q$ are defined on $\mathbb{R}^d$ where $d>1$, both exact transformation and approximation may be hard. Having a large number of layers is a necessary (but not a sufficient) condition.
Challenges
Our problem is very related to the universal approximation property: the ability of a function class to be arbitrarily close to any target function. Although we have this property for shallow neural networks, fully connected networks, and residual networks, these results do not apply to NFs. Why? Because of the invertibility.
First, a function class has the universal approximation property does not imply that its invertible subset can approximate between any pair of distributions. For instance, take the set of piecewise constant functions. Its invertible subset is the empty set!
On the other hand, a function class has limited capacity does not imply that its invertible subset cannot transform between any pair of distributions. For instance, take the set of triangular maps, which can perform powerful Knothe–Rosenblatt rearrangements (See page 17 of this book).
The way to get around this challenge: instead of looking at the capacity of a function class in the function space, we directly analyze input–output distribution pairs.
Universal Approximation When $d=1$
As warm-up let us look at the one-dimensional case. We show planar layers can approximate between arbitrary pairs of distributions under mild assumptions. We analyze a specific kind of planar layer with the ReLU activation:
\[f_{\text{pf}}(z)=z+u\ \mathrm{ReLU}(wz+b)\]
where $u,w,b,z\in\mathbb{R}$, and $\text{ReLU}(x)=\max(x,0)$. The effect of this transformation on a density is first splitting its graph into two pieces, and then scaling one piece while keeping the other one unchanged. For example, in the figure below the first planar layer splits the blue line into the solid part and the dashed part, and scales the dashed part to the orange line. Similarly, the second planar layer splits the orange line into the solid part and the dashed part, and scales the dashed part to the green line.
In particular, if the blue line is Gaussian, then the orange line and the green line are also pieces of some Gaussian distributions. We call this a piecewise Gaussian distribution. Additionally, it has the consistency property: the integration of the transformed distribution should always be 1.
How does it relate to approximation? Here we use a fundamental result in real analysis: Lebesgue-integrable functions can be approximated by piecewise constant functions. Given a piecewise constant distribution $q_{\text{pwc}}$ that is close to the target distribution $p$, we can iteratively construct a piecewise Gaussian distribution $q_{\text{pwg}}$ with the same group of pieces. We can additionally require $q_{\text{pwg}}$ to be very close to $q_{\text{pwc}}$ by carefully selecting the parameters $u,w,b$. Finally, as the pieces become smaller, $q_{\text{pwc}}\rightarrow p$ and $q_{\text{pwg}}\rightarrow q_{\text{pwc}}$, which implies $q_{\text{pwg}}\rightarrow p$.
In the following example, we demonstrate such approximation with 50(top) and 300(bottom) ReLU planar layers, respectively.
Exact Transformation When $d>1$
Next, we look at the more general case in higher-dimensional space, which is usually quite different from the one-dimensional case. We show exact transformation between distributions can be quite hard. Specifically, we analyze Sylvester layers, a matrix-form generalization of planar layers (note that on $\mathbb{R}$, planar layers and Sylvester layers are equivalent):
\[f_{\text{syl}}(z)=z+Ah(B^{\top}z+b)\]
where $A,B\in\mathbb{R}^{d\times m},z\in\mathbb{R}^d,b\in\mathbb{R}^m$ for some integer $m$. In particular, we call $m$ the number of neurons of $f_{\text{syl}}$ because its form is identical to a residual block with $m$ neurons in the hidden layer.
Now suppose we stack a number of Sylvester layers with $M$ neurons in total, and these layers sequentially transform an input distribution $q$ to output distribution $p$. For convenience, let $f$ be the function composed of all these Sylvester layers. We show that the distribution pairs $(q,p)$ must obey some necessary (but not sufficient) condition, which we call the topology matching condition.
$h$ is a smooth function
Let $L(z)=\log p(f(z))-\log q(z)$ be the log-det Jacobian term. Then, the topology matching condition says the dimension of the set of the gradient of $L$ is no more than the number of neurons. Formally,
\[\dim\{\nabla_z L(z):z\in\mathbb{R}^d\}\leq M\]
In other words, if $M$ is less than the above dimensionality then exact transformation is impossible no matter what smooth non-linearities $h$ are selected.
Since it is not easy to plot $\{\nabla_z L(z):z\in\mathbb{R}^d\}$, we demonstrate $L(z)$ in a few examples below. Each row is a group, containing plots of $q$, $p$, and $L$ from left to right. In these examples, $M=1$ so $\nabla_z L(z)$ is a multiple a constant vector.
→
→
→
→
Based on the topology matching condition, it can be shown that if the number of neurons $M$ is less than the dimension $d$, it may even be hard to transform between simple Gaussian distributions.
When $h=\text{ReLU}$
We then restrict to ReLU Sylvester layers. In this case, $f$ in fact performs a piecewise linear transformation in $\mathbb{R}^d$. As a result, for almost every $z\in\mathbb{R}^d$ (except for boundary points), $f$ is linear around $z$. This leads to the following (pointwise) topology matching condition: there exists a constant matrix $C$ (which is the Jacobian matrix of $f(z)$) around $z$ such that
\[C^{\top}\nabla_z\log p(f(z))=\nabla_z\log q(z)\]
We demonstrate this result with two examples below, where each row is a $(q,p)$ distribution pair. The red points ($z$) on the left are transformed to those ($f(z)$) on the right by $f$. Notice that these red points are peaks of $q$ and $p$, respectively. In these cases, both $\nabla_z\log p(f(z))$ and $\nabla_z\log q(z)$ are zero vectors, which is compatible with the topology matching condition.
→
→
As a corollary, we conclude that ReLU Sylvester layers generally do not transform between product distributions or mixture of Gaussian distributions except for very special cases.
Approximation Capacity When $d>1$
It is not surprising that exact transformation between distributions is difficult. What if we loosen our goal to approximation between distributions, where we can use transformations from a certain class $\mathcal{F}$? We show that unfortunately, this is still hard under certain conditions.
The way to look at this problem is to bound the minimum depth that is needed to approximate between $q$ and $p$. In other words, if we use less than this number of transformations, then it is impossible to approximate $p$ given $q$ as the source, no matter what transformations in $\mathcal{F}$ are selected. Formally, for $\epsilon>0$, we define the minimum depth as
\[T_{\epsilon}(p,q,\mathcal{F})=\inf\{n: \exists \{f_i\}_{i=1}^n\in\mathcal{F}\text{ such that }\mathrm{TV}((f_1\circ\cdots\circ f_n)(q),p)\leq\epsilon\}\]
where $\mathrm{TV}$ is the total variance distance.
We conclude that if $\mathcal{F}$ is the set of $(i)$ planar layers $f_{\text{pf}}$ with bounded parameters and popular non-linearities including $\tanh$, sigmoid, and $\arctan$, or $(ii)$ all Householder layers $f_{\text{hh}}$, then $T_{\epsilon}(p,q,\mathcal{F})$ is not small. In detail, for any $\kappa>0$, there exists a pair of distributions $(q,p)$ on $\mathbb{R}^d$ and a constant $\epsilon$ (e.g., 0.5) such that
\[T_{\epsilon}(p,q,\mathcal{F})=\tilde{\Omega}(d^{\kappa})\]
Although this lower bound is polynomial in the dimension $d$, in many practical problems the dimension can be very large so the minimum depth is still an incredibly large number. This result tells us that planar layers and Householder layers are provably not very expressive under certain conditions.
Open Problems
This is the end of our paper, but is clearly just the beginning of the story. There are a large number of open problems on the expressive power of even simple normalizing flow transformations. Below are some potential directions.
Just like neural networks, planar and Sylvester layers use non-linearities in their expressions. Is it possible that a certain combination of non-linearities (at different layers) can significantly improve capacity?
Our paper does not provide a result for very deep Sylvester flows (e.g., $>d$ layers) with smooth non-linearities. Therefore, it is interesting to provide some insights for deep Sylvester flows.
A more general problem is to understand if the universal approximation property of certain class of normalizing flows holds in converting between distributions. The result is meaningful even if we assume the depth can be arbitrarily large.
On the other hand, it is also helpful to analyze what these normalizing flows are good at. A good example is to show that they can easily transform between distributions in a certain class, especially by an elegant construction.
]]><a href='https://cseweb.ucsd.edu/~z4kong'>Zhifeng Kong</a> and <a href='http://cseweb.ucsd.edu/~kamalika'>Kamalika Chaudhuri</a>Explainable 2-means Clustering: Five Lines Proof2020-10-16T18:00:00+00:002020-10-16T18:00:00+00:00https://ucsdml.github.io//jekyll/update/2020/10/16/explain_2_meansTL;DR: we will show why only one feature is enough to define a good $2$-means clustering. And we will do it using only 5 inequalities (!)
In a previous post, we explained what is an explainable clustering.
Explainable clustering
In a previous post, we discussed why explainability is important, defined it as a small decision tree, and suggested an algorithm to find such a clustering. But why the resulting clustering is any good?? We measure “good” by $k$-means cost. The cost of a clustering $C$ is defined as the sum of squared Euclidean distances of each point $x$ to its center $c(x)$. Formally,
\begin{equation}
cost(C)=\sum_x \|x-c(x)\|^2,
\end{equation} the sum is over all points $x$ in the dataset.
In this post, we focus on the $2$-means problem, where there are only two clusters. We want to show that for every dataset there is one feature $i$ and one threshold $\theta$ such that the following simple clustering $C^{i,\theta}=(C^{i,\theta}_1,C^{i,\theta}_2)$ has a low cost:
\begin{equation}
\text{if } x_i\leq\theta \text{ then } x\in C^{i,\theta}_1 \text{ else } x\in C^{i,\theta}_2.
\end{equation}
We call such a clustering a threshold cut. There might be many threshold cuts that are good, bad, or somewhere in between. We want to show that there is at least one that is good (i.e., low cost). In the paper, we prove that there is always a threshold cut, $C^{i,\theta}$, that is almost as good as the optimal clustering:
\begin{equation}
cost(C^{i,\theta})\leq4\cdot cost(opt),
\end{equation}
where $cost(opt)$ is the cost of the optimal 2-means clustering. This means that there is a simple explainable clustering $C^{i,\theta}$ that is only $4$ times worse than the optimal one. It’s independent of the dimension and the number of points. Sounds crazy, right? Let’s see how we can prove it!
The minimal-mistakes threshold cut
We want to compare two clusterings: the optimal clustering and the best threshold cut. The best threshold cut is hard to analyze, so we introduce an intermediate clustering: the minimal-mistakes threshold cut, $\widehat{C}$. Even though this clustering will not be the best threshold cut, it will be good enough. In the paper we prove that $cost(\widehat{C})$ is at most $4cost(opt)$. For simplicity, in this post, we will show a slightly worse bound of $11cost(opt)$ instead of $4cost(opt)$.
We define the number of mistakes of a threshold cut $C^{i,\theta}$ as the number of points $x$ that are not in the same cluster as their optimal center $c(x)$ in $C^{i,\theta}$, i.e., number of points $x$ such that
\begin{equation}
sign(\theta-x_i) \neq sign(\theta-c(x)_i).
\end{equation}
The minimal-mistakes clustering is the threshold cut that has the minimal number of mistakes. Take a look at the next figure for an example.
Two optimal clusters are in red and blue. Centers are the stars. Split (in yellow) with one mistake. This is a minimal-mistakes threshold cut, as any threshold cut has at least $1$ mistake.
Playing with cost: warm-up
Before we present the proof, let’s familiarize ourselves with the $k$-means cost and explore several of its properties. It will be helpful later on!
Changing centers
If we change the centers of a clustering from their means (which are their optimal centers) to different centers $c=(c_1, c_2)$, then the cost can only increase. Putting this into math, denote by $cost(C,c)$ the cost of clustering $C=(C_1,C_2)$ when $c_1$ is the center of cluster $C_1$ and $c_2$ is the center of cluster $C_2$, then
\begin{align}
cost(C) &= \sum_{x\in C_1} \|x-mean(C_1)\|^2 + \sum_{x\in C_2} \|x-mean(C_2)\|^2 \newline &\leq \sum_{x\in C_1} \|x-c_1\|^2 + \sum_{x\in C_2} \|x-c_2\|^2 = cost(C,c).
\end{align}
What if we further want to change the centers from some arbitrary centers $(c_1, c_2)$ to other arbitrary centers $(m_1, m_2)$? How does the cost change? Can we bound it? To our rescue comes the (almost) triangle inequality that states that for any two vectors $x,y$:
\begin{equation}
\|x+y\|^2 \leq 2\|x\|^2+2\|y\|^2.
\end{equation}
This implies that the cost of changing the centers from $c=(c_1, c_2)$ to $m=(m_1, m_2)$ is bounded by
\begin{equation}
cost(C,c)\leq 2cost(C,m)+2|C_1|\|c_1-m_1\|^2+2|C_2|\|c_2-m_2\|^2.
\end{equation}
Decomposing the cost
The cost can be easily decomposed with respect to the data points and the features. Let’s start with the data points. For any partition of the points in $C$ to $S_1$ and $S_2$, the cost can be rewritten as
\begin{equation}
cost(C,c)=cost(C \cap S_1,c)+cost(C \cap S_2,c).
\end{equation}
The cost can also be decomposed with respect to the features, because we are using the squared Euclidean distance. To be more specific, the cost incur by the $i$-th feature is $cost_i(C,c)=\sum_{x}(x_i-c(x)_i)^2,$ and the total cost is equal to
\begin{equation}
cost(C,c)=\sum_i cost_i(C,c).
\end{equation}
If the last equation is unclear just recall the definition of the cost ($c(x$) is the center of a point $x$):
\begin{equation}
cost(C,c)=\sum_{x}\|x-c(x)\|^2=\sum_i\sum_{x}(x_i-c(x)_i)^2=\sum_icost_i(C,c).
\end{equation}
The 5-line proof
Now we are ready to prove that $\widehat{C}$ is only a constant factor worse than the optimal $2$-means clustering:
\begin{equation}
cost(\widehat{C})\leq 11\cdot cost(opt).
\end{equation}
To prove that the minimal-mistakes threshold cut $\widehat{C}$ gives a low-cost clustering, we will do something that might look strange at first. We analyze the quality of this clustering $\widehat{C}$ with the optimal centers of the optimal clustering. And not the optimal centers for $\widehat{C}$. This step will only increase the cost, so why are we doing it — because it will ease our analysis, and if there are not many mistakes, then the centers do not change much, like in the previous figure. So it’s not much of an increase. So, here comes the first step — change the centers of $\widehat{C}$ to the optimal centers $c^*=(mean(C^*_1),mean(C^*_2))$. Recall from the warm-up that this can only increase the cost:
\begin{equation}
cost(\widehat{C})\leq cost(\widehat{C},c^{*}) \quad (1)
\end{equation}
Next we use one of the decomposition properties of the cost. We partition the dataset into the set of points that are correctly labeled, $X^{cor}$, and those that are not, $X^{wro}$.
The same dataset and split as before. Point with a grey circle is in the wrong cluster and is the only member in $X^{wro}$. All other points have the same cluster assignment as the optimal clustering and are in $X^{cor}$.
Thus, we can rewrite the last term as
\begin{equation}
cost(\widehat{C},c^{*})=cost(\widehat{C}\cap X^{cor},c^{*})+cost(\widehat{C}\cap X^{wro},c^{*}) \quad (2)
\end{equation}
Let’s look at this sum. The first term contains all the points that have their correct center in $c^*$ (which is either $mean(C^*_1)$ or $mean(C^*_2)$). Hence, the first term in (2) is easy to bound: it’s at most $cost(opt)$. So from now on, we focus on the second term.
In the second term, all points are in $X^{wro}$, which means they were assigned to the incorrect optimal center. So let’s change the centers once more, so that $X^{wro}$ will have the correct centers. The correct centers of $X^{wro}$ are the same centers $c^*$, but the order is reversed, i.e., all points assigned to center $mean(C^*_1)$ are now assigned to $mean(C^*_2)$ and vice versa. Using the “changing centers” property of the cost we discussed earlier, we have
Now we’ve reached the main step in the proof. We show that the second term in (3) is bounded by $8cost(opt)$. We first decompose $cost(opt)$ using the features. Then, all we need to show is that:
The trick is, for each feature, to focus on the threshold cut defined by the middle point between the two optimal centers. Since $\widehat{C}$ is the minimal-mistakes clustering we know that in every threshold cut there are at least $|X^{wro}|$ mistakes. Each mistake contributes at least half the distance between the two centers.
Proving step $4.$ Projecting to feature $i$. Points in blue belong to the first cluster, and in red to the second. We focus on the cut that is the mid-point between the two optimal centers.
This figure shows how to prove step (4). We see that there is $1$ mistake, which is the minimum possible. This means that even the optimal clustering must pay for at least half the distance between the centers for each of these mistakes. This gives us a lower bound on $cost_i(opt)$ in this feature. Then we can sum over all the features to see that the second term of (3) is at most $8cost(opt)$, which is what we wanted.
Putting everything together, we get exactly what we wanted to prove in this post:
\begin{equation}
cost(\widehat{C})\leq1 1\cdot cost(opt) \quad (5)
\end{equation}
Epilogue: improvements
The bound that we got, $11$, is not the best possible. With more tricks we can get a bound of $4$. One of them is using Hall’s theorem. Similar ideas provide a $2$-approximation to the optimal $2$-medians clustering as well.
To complement our upper bounds, we also prove lower bounds showing that any threshold cut must incur almost $3$-approximation for $2$-means and almost $2$-approximation for $2$-medians. You can read all about it in our paper.
]]><a href='https://sites.google.com/view/michal-moshkovitz'>Michal Moshkovitz</a>, <a href='mailto:navefrost@mail.tau.ac.il'>Nave Frost</a>, <a href='https://sites.google.com/site/cyrusrashtchian/'>Cyrus Rashtchian</a>Explainable k-means Clustering2020-10-16T17:00:00+00:002020-10-16T17:00:00+00:00https://ucsdml.github.io//jekyll/update/2020/10/16/explain_k_meansTL;DR:
Explainable AI has gained a lot of interest in the last few years, but effective methods for unsupervised learning are scarce. And the rare methods that do exist do not have provable guarantees. We present a new algorithm for explainable clustering that is provably good for $k$-means clustering — the Iterative Mistake Minimization (IMM) algorithm. Specifically, we want to build a clustering defined by a small decision tree. Overall, this post summarizes our new paper: Explainable $k$-Means and $k$-Medians clustering.
Explainability: why?
Machine learning models are mostly “black box”. They give good results, but their reasoning is unclear. These days, machine learning is entering fields like healthcare (e.g., for a better understanding of Alzheimer’s Disease and Breast Cancer), transportation, or law. In these fields, quality is not the only objective. No matter how well a computer is making its predictions, we can’t even imagine blindly following computer’s suggestion. Can you imagine blindly medicating or performing a surgery on a patient just because a computer said so? Instead, it would be much better to provide insight into what parts of the data the algorithm used to make its prediction.
Tree-based explainable clustering
We study a prominent problem in unsupervised learning, $k$-means clustering. We are given a dataset, and the goal is to partition it to $k$ clusters such that the $k$-means cost is minimal. The cost of a clustering $C=(C^1,\ldots,C^k)$ is the sum of all points from their optimal centers, $mean(C^i)$:
For any cluster, $C^i$, one possible explanation of this cluster is $mean(C^i)$. In a low-cost clustering, the center is close to its points, and they are close to each other. For example, see the next figure.
Near optimal 5-means clustering
Unfortunately, this explanation is not as useful as it could be. The centers themselves may depend on all the data points and all the features in a complicated way. We instead aim to develop a clustering method that is explainable by design. To explain why a point is in a cluster, we will only need to look at small number of features, and we will just evaluate a threshold for each feature one by one. This allows us to extract information about which features cause a point to go to one cluster compared to another. This method also means that we can derive an explanation that does not depend on the centers.
More formally, at each step we test if $x_i\leq \theta$ or not, for some feature $i$ and threshold $\theta$. We call this test a split. According to the test’s result, we decide on the next step. In the end, the algorithm returns the cluster identity. This procedure is exactly a decision tree where the leaves correspond to clusters.
Importantly, for the tree to be explainable it should be small. The smallest decision tree has $k$ leaves since each cluster must appear in at least one leaf. We call a clustering defined by a decision tree with $k$ leaves a tree-based explainable clustering. See the next tree for an illustration.
On the left, we see a decision tree that defines a clustering with $5$ clusters. On the right, we see the geometric representation of this decision tree. We see that the decision tree imposes a partition to $5$ clusters aligned to the axis. The clustering looks close to the optimal clustering that we started with. Which is great. But can we do it for all datasets? How?
Several algorithms are trying to find a tree-based explainable clustering like CLTree and CUBT. But we are the first to give formal guarantees. We first need to define the quality of an algorithm. It’s common that unsupervised learning problems are NP-hard. Clustering is no exception. So it is common to settle for an approximated solution. A bit more formal, an algorithm that returns a tree-based clustering $T$ is an $a$-approximation if $cost(T)\leq a\cdot cost(opt),$ where $opt$ is the clustering that minimizes the $k$-means cost.
General scheme
Many supervised learning algorithms learn a decision tree, can we use one of them here? Yes, after we transform the problem into a supervised learning problem! How might you ask? We can use any clustering algorithm that will return a good, but not explainable clustering. This will form the labeling. Next, we can use a supervised algorithm that learns a decision tree. Let’s summarize these three steps:
Find a clustering using some clustering algorithm
Label each example according to its cluster
Call a supervised algorithm that learns a decision tree
Which algorithm can we use in step 3? Maybe the popular ID3 algorithm?
Can we use the ID3 algorithm?
Short answer: no.
One might hope that in step 3, in the previous scheme, the known ID3 algorithm can be used (or one of its variants like C4.5). We will show that this does not work. There are datasets where ID3 will perform poorly. Here is an example:
ID3 performs poorly on this dataset
The dataset is composed of three clusters, as you can see in the figure above. Two large clusters (0 and 1 in the figure) have centers (-2, 0) and (2, 0) accordingly and small noise. The third cluster (2 in the figure) is composed of only two points that are very, very (very) far away from clusters 0 and 1. Given these data, ID3 will prefer to maximize the information gain and split between clusters 0 and 1. Recall that the final tree has only three leaves. This means that in the final tree, one point in cluster 2 must be with cluster 0 or cluster 1. Thus the cost is enormous.
To solve this problem, we design a new algorithm called Iterative Mistake Minimization (IMM).
IMM algorithm for explainable clustering
We learned that the ID3 algorithm cannot be used in step 3 at the general scheme. Before we give up on this scheme, can we use a different decision-tree algorithm? Well, since we wrote this post, you probably know the answer: there is such an algorithm, the IMM algorithm.
We build the tree greedily from top to bottom. Each step we take the split (i.e., feature and threshold) that minimizes a new parameter called a mistake. A point $x$ is a mistake for node $u$ if $x$ and its center $c(x)$ reached $u$ and then separated by $u$’s split. See the next figure for an example of a split with one mistake.
Split (in yellow) with one mistake. Two optimal clusters are in red and blue. Centers are the stars.
To summarize, the high-level description of the IMM algorithm:
As long as there is more than one center
find the split with minimal number of mistakes
Here is an illustration of the IMM algorithm. We use $k$-means++ with $k=5$ to find a clustering for our dataset. Each point is colored with its cluster label. At each node in the tree, we choose a split with a minimal number of mistakes. We stop where each of the $k=5$ centers is in its own leaf. This defines the explainable clustering on the left.
The algorithm is guaranteed to perform well. For any dataset. See the next theorem.
IMM is an $O(k^2)$-approximation to the optimal $k$-means clustering.
This theorem shows that we can always find a small tree, with $k$ leaves, such that the tree-based clustering is only $O(k^2)$ times worse in terms of the cost. IMM efficiently find this explainable clustering. Importantly, this approximation is independent of the dimension and the number of points. A proof for the case $k=2$ will appear in a follow-up post, and you can read the proof for general $k$ in the paper. Intuitively, we discovered that the number of mistakes is a good indicator for the $k$-means cost, and so, minimizing the number of mistakes is an effective way to find a low-cost clustering.
Running Time
What is the running time of the IMM algorithm? With an efficient implementation, using dynamic programming, the running time is $O(kdn\log(n)).$ Why? For each of the $k-1$ inner nodes and each of the $d$ features, we can find the split that minimizes the number of mistakes for this node and feature, in time $O(n\log(n)).$
For $2$-means one can do better than running IMM: going over all possible $(n-1)d$ cuts and find the best one. The running time is $O(nd^2+nd\log(n))$.
Results Summary
In each cell in the following table, we write the approximation factor. We want this value to be small for the upper bounds and large for the lower bounds. In $2$-medians, the upper and lower bounds are pretty tight, about $2$. But, there is a large gap for $k$-means and $k$-median: the lower bound is $\log(k)$, while the upper bound is $\mathsf{poly}(k)$.
$k$-medians
$k$-means
$k=2$
$k>2$
$k=2$
$k>2$
Lower
$2-\frac1d$
$\Omega(\log k)$
$3\left(1-\frac1d\right)^2$
$\Omega(\log k)$
Upper
$2$
$O(k)$
$4$
$O(k^2)$
What’s next
IMM exhibits excellent results in practice on many datasets, see this. It’s running time is comparable to KMeans implemented in sklearn. We implemented the IMM algorithm, it’s here. Try it yourself.
We plan to have several posts on explainable clusterings, here is the second in the series, stay tuned for more!
In a follow-up work, we explore the tradeoff between explainability and accuracy. If we allow a slightly larger tree, can we get a lower cost? We introduce the ExKMC, “Expanding Explainable $k$-Means Clustering”, algorithm that builds on IMM.
Found cool applications of IMM? Let us know!
]]><a href='https://sites.google.com/view/michal-moshkovitz'>Michal Moshkovitz</a>, <a href='mailto:navefrost@mail.tau.ac.il'>Nave Frost</a>, <a href='https://sites.google.com/site/cyrusrashtchian/'>Cyrus Rashtchian</a>Towards Physics-informed Deep Learning for Turbulent Flow Prediction2020-08-23T00:00:00+00:002020-08-23T00:00:00+00:00https://ucsdml.github.io//jekyll/update/2020/08/23/TF-NetPrediction Visualization
We propose a novel hybrid model for turbulence prediction, $\texttt{TF-Net}$, that unifies a popular Computational fluid dynamics (CFD) technique, RANS-LES coupling, with custom-designed U-net. The following two videos show the ground truth and the predicted U (left) and V (right) velocity fields from $\texttt{TF-Net}$ and three best baselines. We see that the predictions by $\texttt{TF-Net}$ are the closest to the target based on the shape and the frequency of the motions. Baselines generate smooth predictions and miss the details of small scale motion.
Introduction
Modeling the spatiotemporal dynamics over a wide range of space and time scales is a fundamental task in science, especially atmospheric science, marine science and aerodynamics. Computational fluid dynamics (CFD) is at the heart of climate modeling and has direct implications for understanding and predicting climate change. Recently, deep learning have demonstrated great success in the automation, acceleration, and streamlining of highly compute-intensive workflows for science. We hope deep learning can accelerate the turbulence simulation since the current CFD is purely physics-based and computationally-intensive, requiring significant computational resources and expertise.
But purely data-driven methods are mainly statistical with no underlying physical knowledge incorporated, and are yet to be proven to be successful in capturing and predicting accurately the complex physical systems. Incorporating physics knowledge into deep learning models can improve not only prediction accuracy, but more importantly, physical consistency. Thus, developing deep learning methods that can incorporate physical laws in a systematic manner is a key element in advancing AI for physical sciences.
Computational techniques are at the core of present-day turbulence investigations, which are a branch of fluid mechanics that uses numerical method to analyze and predict fluid flows. In physics, people use the following Navier–Stokes equations to describe the motion of viscous fluid dynamics.
where $\pmb{w}(t)$ is the vector velocity field of the flow, which is what we want to predict. $p$ and $T$ are pressure and temperature respectively, $\kappa$ is the coefficient of heat conductivity, $\rho_0$ is density at temperature at the beginning, $\alpha$ is the coefficient of thermal expansion, $\nu$ is the kinematic viscosity, $f$ the body force that is due to gravity.
Turbulent-Flow Net
For turbulent flows, the range of length scales and complexity of phenomena involved in turbulence make Direct Numerical Simulation (DNS) approaches prohibitively expensive. Great emphasis was then placed on the alternative approaches including Large-Eddy Simulation (LES), Reynolds-averaged Navier Stokes (RANS) as well as Hybrid RANS-LES Coupling that combines both RANS and LES approaches in order to take advantage of both methods. These methods decompose the fluid flow into different scales in order to directly simulate large scales while model small ones.
Hybrid RANS-LES Coupling decomposes the flow velocity into three scales: mean flow, resolved fluctuations and unresolved fluctuations. It applies the spatial filtering operator $S$ and the temporal average operator $T$ sequentially.
\[\pmb{w^*}(\pmb{x},t) = S \ast\pmb{w} = \sum_{\pmb{\xi}} S(\pmb{x}|\pmb{\xi})\pmb{w}(\pmb{\xi},t)\]
\[\pmb{\bar{w}}(\pmb{x},t) = T \ast \pmb{w^*} = \frac{1}{n}\sum_{s = t-n}^tT(s) \pmb{w^*} (\pmb{x}, s)\]
then $\pmb{\tilde{w}}$ can be defined as the difference between $\pmb{w^*}$ and $\pmb{\bar{w}}$:
The figure below shows this three-level decomposition in wavenumber space. $k$ is the wavenumber, the spatial frequency in the Fourier domain. $E(k)$ is the energy spectrum describing how much kinetic energy is contained in eddies with wavenumber $k$. Small $k$ corresponds to large eddies that contain most of the energy. The slope of the spectrum is negative and indicates the transfer of energy from large scales of motion to the small scales.
Inspired by the hybrid RANS-LES Coupling, we propose a hybrid deep learning framework, $\texttt{TF-Net}$, based on the multilevel spectral decomposition. Specifically, we decompose the velocity field into three scales using the spatial filter $S$ and the temporal filter $T$. Unlike traditional CFD, both filters in $\texttt{TF-Net}$ are trainable neural networks. The motivation for this design is to explicitly guide the DL model to learn the non-linear dynamics of both large and small eddies. We design three identical convolutional encoders to encode the three scale components separately and use a shared convolutional decoder to learn the interactions among these three components and generate the final prediction. The figure below shows the overall architecture of our hybrid model $\texttt{TF-Net}$.
Since the turbulent flow under investigation has zero divergence, we include $\Vert\nabla \cdot \pmb{w}\Vert^2$ as a regularizer to constrain the predictions, leading to a constrained TF-Net, $\texttt{Con TF-Net}$.
Results
We compare our model with four purely data-driven deep learning models, including $\texttt{ResNet}$, $\texttt{ConvLSTM}$, $\texttt{U-net}$ and $\texttt{GAN}$, and two hybrid physics-informed models, including $\texttt{DHPM}$ and $\texttt{SST}$. All the models trained to make one step ahead prediction given the historic frames and we use them autoregressively to generate multi-step forecasts.
$\textbf{Accuracy}$ The following figure show the growth of RMSE with prediction horizon up to 60 time steps ahead. We can see that $\texttt{TF-Net}$ consistently outperforms all baselines, and constraining it with divergence free regularizer can further improve the performance.
$\textbf{Physical Consistency}$ The left figure below is the averages of absolute divergence over all pixels at each prediction step and the right figure below is the energy spectrum curves. $\texttt{TF-Net}$ predictions are in fact much closer to the target even without additional divergence free constraint, which suggests that $\texttt{TF-Net}$ can generate predictions that are physically consistent with the ground truth.
$\textbf{Efficiency}$ This figure shows the average time to produce one 64 × 448 2d velocity field for all models on single V100 GPU. We can see that $\texttt{TF-net}$, $\texttt{U_net}$ and $\texttt{GAN}$ are faster than the numerical Lattice Boltzmann method. $\texttt{TF-Net}$ will show greater advantage of speed on higher resolution data.
$\textbf{Ablation Study}$ We also perform an ablation study to understand each component of $\texttt{TF-Net}$ and investigate whether the model has actually learned the flow with different scales. During inference, we applied each small U-net in $\texttt{TF-Net}$ with the other two encoders removed to the entire input domain. The video below includes $\texttt{TF-Net}$ predictions and the outputs of each small $\texttt{U-net}$ while the other two encoders are zeroed out. We observe that the outputs of each small $\texttt{U-net}$ are the flow with different scales, which demonstrates that $\texttt{TF-Net}$ can learn multi-scale behaviors.
Conclusion and Future Work
We presented a novel hybrid deep learning model, $\texttt{TF-Net}$, that unifies representation learning and turbulence simulation techniques. $\texttt{TF-Net}$ exploits the multi-scale behavior of turbulent flows to design trainable scale-separation operators to model different ranges of scales individually. We provide exhaustive comparisons of $\texttt{TF-Net}$ and baselines and observe significant improvement in both the prediction error and desired physical quantifies, including divergence, turbulence kinetic energy and energy spectrum. Future work includes extending these techniques to very high-resolution, 3D turbulent flows and incorporating additional physical variables, such as pressure and temperature, and additional physical constraints, such as conservation of momentum, to improve the accuracy and faithfulness of deep learning models.